标签:level app extract tables 生物 include reads des marker
---恢复内容开始---
上周20161021-20161028的任务还没有搞完,所以今天来填坑(微笑脸)
××××××××××××××××××××我是萌萌哒分割线×××××××××××××××××××××××××××××××
本文参考:https://bitbucket.org/biobakery/biobakery/wiki/metaphlan2
MetaPhlAn2是使用宏基因组鸟枪法测序数据描绘微生物群落组成的工具。

ps: 与系统发育相关的图的表示

安装:
1. 下载并解压 2. 修改/etc/profile,添加路径 3. 直接使用metaphlan2.py
4. 使用这种命令时要给出bowtie2的路径--bowtie2_exe<bowtie2>.
××××××××××××××××××××××××××啦啦啦××××××××××××××××××××××××××××
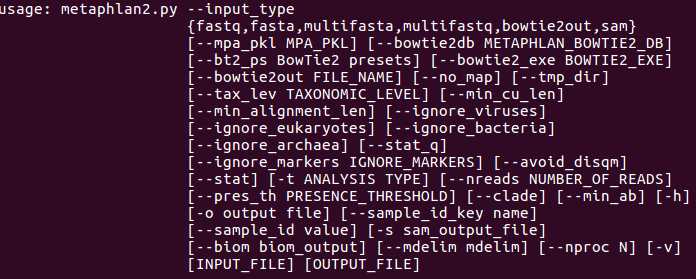
帮助信息:metaphlan2.py -h | less使用上下箭头滚动查看,q退出。
实验内容:
Step 0 : 下载数据(右键,copy link address)
mkdir mytest
cd mytest
wget https://bitbucket.org/biobakery/biobakery/raw/tip/demos/biobakery_demos/data/metaphlan2/input/SRS014459-Stool.fasta.gz
Step 1: 使用单个reads文件作为输入
metaphlan2.py SRS014476-Supragingival_plaque.fasta.gz --input_type fasta > SRS014476-Supragingival_plaque_profile.txt
bowtie2out.txt: intermediate mapping results to unique sequence markers
可以用这个命令查看(不过我觉得用gedit也可以啊==除非是命令行)
less -S SRS014476-Supragingival_plaque.fasta.gz.bowtie2out.txt
profile.txt:最终的结果
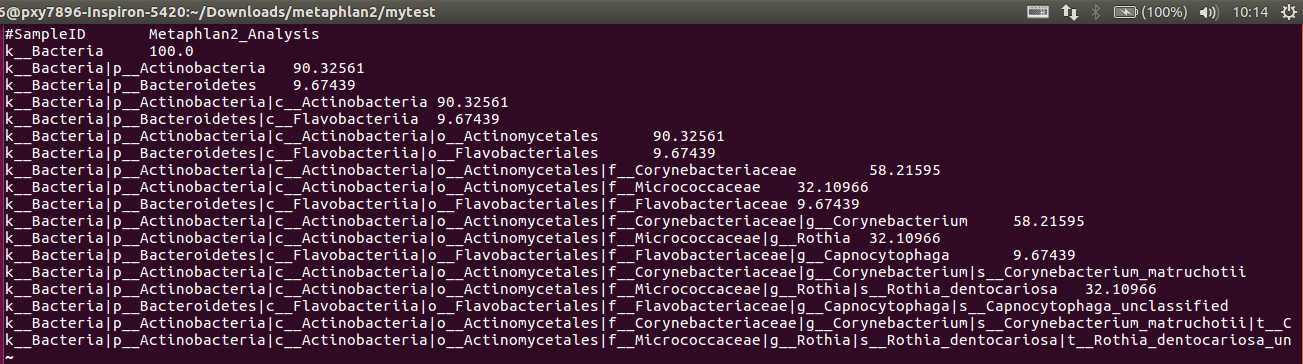
解释:
1. 这就是对分类的描述:Kingdom: k__, Phylum: p__, Class: c__, Order: o__, Family: f__, Genus: g__, Species: s__
2. 数字是百分比
3. OTU equivalents can be extracted by using only the species-level s__ clades from this file (again, making
sure to include clades unclassified at this level).
Step 2: 多线程处理--nproc命令
metaphlan2.py SRS014459-Stool.fasta.gz --input_type fasta --nproc 4 > SRS014459-Stool_profile.txt
Step 3: 多输入处理
for f in *.fasta.gz
do
metaphlan2.py $f --input_type fasta --nproc 4 > ${f%.fasta.gz}_profile.txt
done
Step 4: 合并结果(使用utils/merge_metaphlan_tables.py脚本)
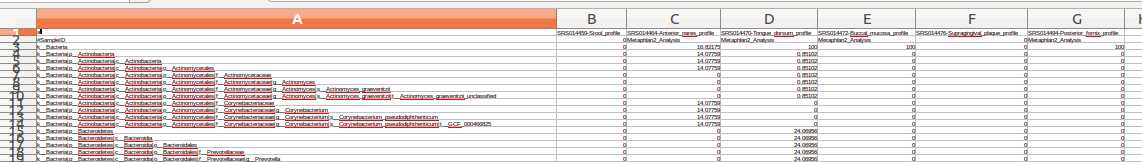
/home/pxy7896/Downloads/metaphlan2/utils/merge_metaphlan_tables.py *_profile.txt > merged_abundance_table.txt
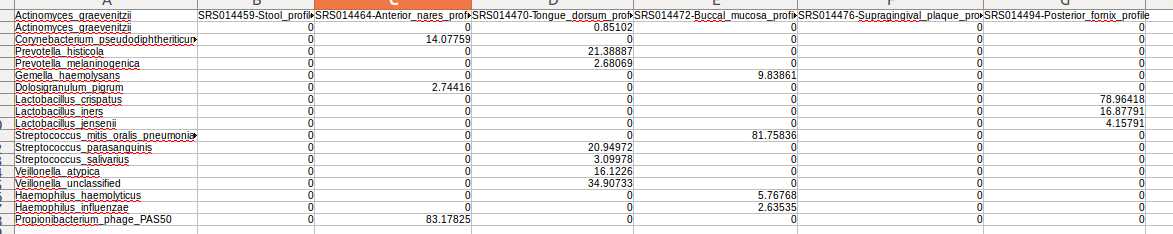
可以用LibreOffice Calc查看。。。因为数据多,有点丑。大概如下图,前面是分类信息,后面是几个文件的数量信息。
Step 5: 数据可视化,heatmap
(1) 安装brew
sudo apt install linuxbrew-wrapper
(2) 安装hclust2
brew tap biobakery/biobakery
brew install hclust2
安装失败。。。问题诡异。。。所以我决定先安装R语言包,用里面的函数heatmap什么的来画。
安装R参见:
(3) 产生物种species丰度表(只有物种层面的)
使用命令:grep -E "(s__)|(^ID)" merged_abundance_table.txt | grep -v "t__" | sed ‘s/^.*s__//g‘ > merged_abundance_table_species.txt
命令解释:
1. grep是按指定模式输出的一个软件(库),参考用户手册:http://www.gnu.org/software/grep/manual/grep.pdf
2.

标签:level app extract tables 生物 include reads des marker
原文地址:http://www.cnblogs.com/pxy7896/p/6012871.html