标签:
用途:trim, crop, remove adapters
支持gz, bz2的fastq
http://www.usadellab.org/cms/?page=trimmomatic
http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf
先下载二进制包
http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/Trimmomatic-0.33.zip
然后解压unzip
我有写MP文库,我的目的是将read开头第一个碱基的N去掉,如果第六个碱基还是N, 那前六个都去掉,去掉后最后保留前40个碱基
ILLUMINACLIP: Cut adapter and other illumina-specific sequences from the read.
SLIDINGWINDOW: Performs a sliding window trimming approach. It starts scanning at the 5? end and clips the read once the average quality within the window falls below a threshold.
MAXINFO: An adaptive quality trimmer which balances read length and error rate to maximise the value of each read
LEADING: Cut bases off the start of a read, if below a threshold quality
TRAILING: Cut bases off the end of a read, if below a threshold quality
CROP: Cut the read to a specified length by removing bases from the end
HEADCROP: Cut the specified number of bases from the start of the read
MINLEN: Drop the read if it is below a specified length
AVGQUAL: Drop the read if the average quality is below the specified level
TOPHRED33: Convert quality scores to Phred-33
TOPHRED64: Convert quality scores to Phred-64
解压后直接就可以用了
从0.32版本,软件会自动识别是33还是64.
Processing Order The different processing steps occur in the order in which the steps are specified on the command line. It is recommended in most cases that adapter clipping, if required, is done as early as possible, since correctly identifying adapters using partial matches is more difficult.
我不需要去接头,因为对于MP文库,接头在后面,
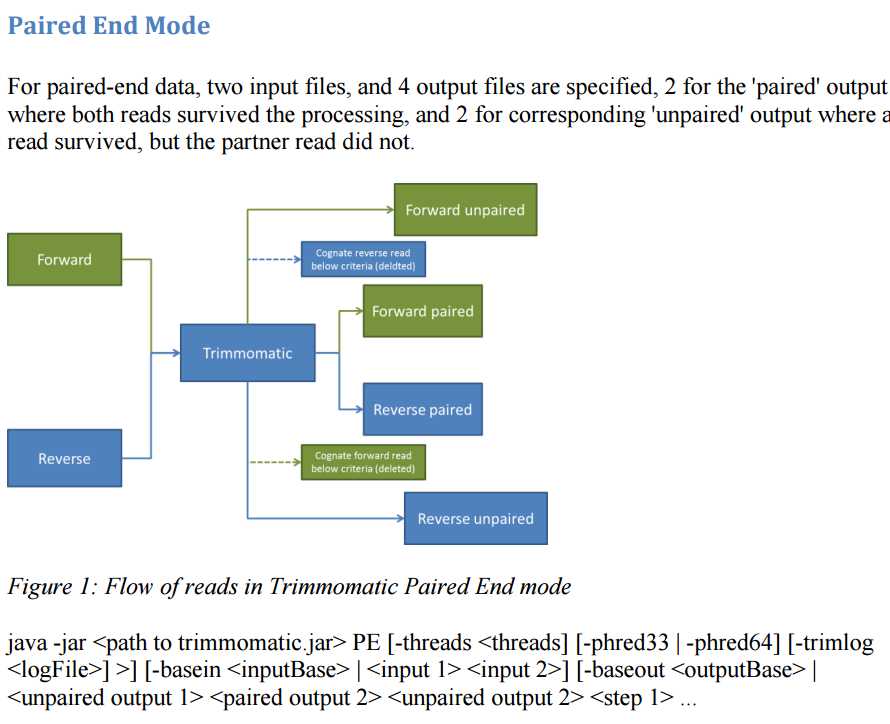
输入和输出的名字要起好:
For input files,
either of the following can be used:
Explicitly naming the 2 input files Naming the forward file using the -basein flag, where the reverse file can be determined automatically.
The second file is determined by looking for common patterns of file naming, and changing the appropriate character to reference the reverse file.
Examples which should be correctly handled include:
Sample_Name_R1_001.fq.gh -> Sample_Name_R2_001.fq.gz
Sample_Name.f.fastq -> Sample_Name.r.fastq
Sample_Name.1.sequence.txt -> Sample_Name.2.sequence.txt
For output files, either of the following can be used:
Explicity naming the 4 output files Providing a base file name using the –baseout flag,
from which the 4 output files can be derived. If the name “mySampleFiltered.fq.gz” is provided, the following 4 file names will be used:
mySampleFiltered_1P.fq.gz - for paired forward reads
mySampleFiltered_1U.fq.gz - for unpaired forward reads
mySampleFiltered_2P.fq.gz - for paired reverse reads
mySampleFiltered_2U.fq.gz - for unpaired reverse reads
Most processing steps take one or more settings, delimited by ‘:‘ (a colon)
SLIDINGWINDOW
Perform a sliding window trimming, cutting once the average quality within the window falls
below a threshold. By considering multiple bases, a single poor quality base will not cause the
removal of high quality data later in the read.
SLIDINGWINDOW:<windowSize>:<requiredQuality>
windowSize: specifies the number of bases to average across
requiredQuality: specifies the average quality required.
The SLIDINGWINDOW trimmer will cut the leftmost position in the window where the average quality drops below the threshold and remove the rest of the read. However if there is low quality in the very beginning of the read then it will fail the minimum length tests and be removed completely - the remaining 3-prime end (even if it is good quality will not be printed)
Consider the following test file t.fq:
@1\1AATGATCGTAGCGATGCAAGCTAGCCCGATGCCCGATCGCATCG+eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeEFCB@2\1AATGATCGTAGCGATGCAAGCTAGCCCGATGCCCGATCGCATCG+EFCBeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee
and processing with:
$ java -jar trimmomatic.jar SE -phred64 t.fq tt.fq SLIDINGWINDOW:4:15TrimmomaticSE: Started with arguments: -phred64 t.fq tt.fq SLIDINGWINDOW:4:15 Automatically using 16 threads Input Reads: 2 Surviving: 1 (50.00%) Dropped: 1 (50.00%) TrimmomaticSE: Completed successfully
The output file looks like the following:
@1\1AATGATCGTAGCGATGCAAGCTAGCCCGATGCCCGATCGC+eeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeeee
As you can see the read with the poor quality at the beginning has been removed completely
LEADING
Remove low quality bases from the beginning. As long as a base has a value below this
threshold the base is removed and the next base will be investigated.
LEADING:<quality>
quality: Specifies the minimum quality required to keep a base.
TRAILING
Remove low quality bases from the end. As long as a base has a value below this threshold
the base is removed and the next base (which as trimmomatic is starting from the 3? prime end
would be base preceding the just removed base) will be investigated. This approach can be
used removing the special illumina „low quality segment? regions (which are marked with
quality score of 2), but we recommend Sliding Window or MaxInfo instead
TRAILING:<quality>
quality: Specifies the minimum quality required to keep a base.
CROP
Removes bases regardless of quality from the end of the read, so that the read has maximally
the specified length after this step has been performed. Steps performed after CROP might of
course further shorten the read.
CROP:<length>
length: The number of bases to keep, from the start of the read.
HEADCROP
Removes the specified number of bases, regardless of quality, from the beginning of the read.
HEADCROP:<length>
length: The number of bases to remove from the start of the read.
Paired End:
java -jar trimmomatic-0.30.jar PE s_1_1_sequence.txt.gz s_1_2_sequence.txt.gz
lane1_forward_paired.fq.gz lane1_forward_unpaired.fq.gz lane1_reverse_paired.fq.gz
lane1_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3
TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36
This will perform the following in this order
1,Remove Illumina adapters provided in the TruSeq3-PE.fa file (provided). Initially Trimmomatic will look for seed matches (16 bases) allowing maximally 2 mismatches. These seeds will be extended and clipped if in the case of paired end reads a score of 30 is reached (about 50 bases), or in the case of single ended reads a score of 10, (about 17 bases).
2,Remove leading low quality or N bases (below quality 3)
3,Remove trailing low quality or N bases (below quality 3)
4,Scan the read with a 4-base wide sliding window, cutting when the average quality per base drops below 15
5, Drop reads which are less than 36 bases long after these steps
最后我的命令如下:
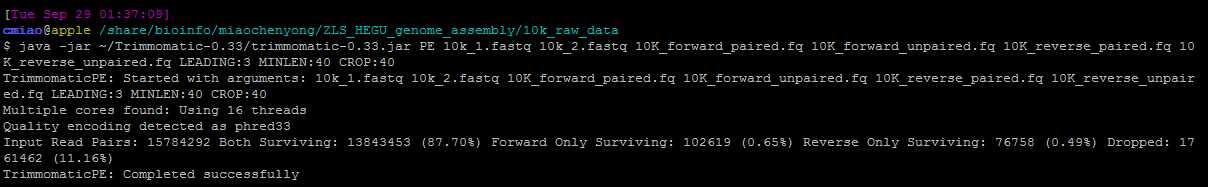
先把开头N去掉,然后前40个碱基,缺点是有些第六个碱基是N的,没法去掉! 估计要写脚本,但是脚本太慢了。。。改天问问老师有没有什么好工具!
freemao
FAFU
标签:
原文地址:http://www.cnblogs.com/freemao/p/4845379.html