标签:
简单好用
identify repeats, to align ESTs and proteins to the genome,
and to automatically synthesize these data into feature-rich gene annotations, including alternative splicing and UTRs, as well as attributes such as evidence trails, and confidence measures.
easily configurable and trainable
its output formats must be both comprehensive and database ready.
provide an easy means to annotate, view, and edit individual contigs and BACs. This allows users to analyze partial genome assemblies and to independently annotate regions of interest using their own data sets, ideally without the overhead of a database and with only minimal compute resources such as a laptop computer.
MAKER identifies repeats, aligns ESTs and proteins to a genome, makes gene predictions, and integrates these data into protein-coding gene annotations. Moreover, its outputs can be loaded directly into GMOD browsers and databases with no post-processing.
MAKER is not exhaustive: it does not identify noncoding RNA genes, nor is it intended as a comprehensive solution to every problem in genome annotation. Rather, MAKER is designed to jump-start genomics in emerging model organisms by providing a robust first round of database-ready protein-coding gene annotations.
We used MAKER on the genomes of both an established and an emerging model organism. Our results for the C. elegans genome demonstrate that the accuracy of MAKER on a model organism genome is comparable to that of other annotation pipelines, whereas our work on the S. mediterranea genome shows that MAKER provides an effective means to annotate an emerging genome and to create a genome database.
MAKER can also be used to annotate individual contigs and BACs.
maker的结构:
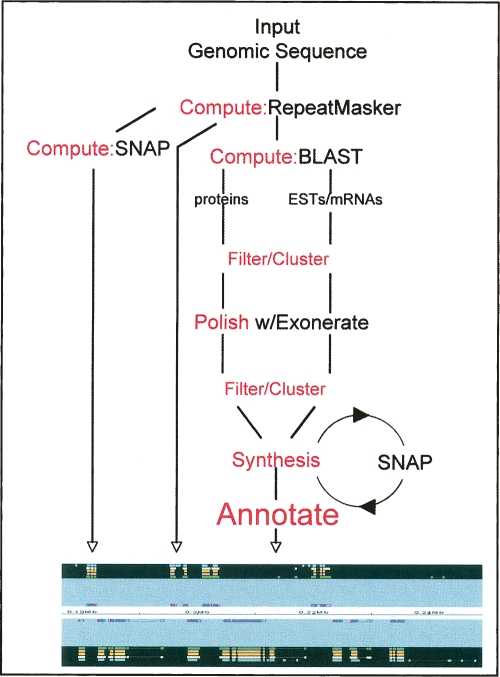
MAKER Overview. MAKER uses four external executables: RepeatMasker, BLAST, SNAP, and Exonerate. Actions corresponding to the five basic steps of automatic annotation are shown in red.
A battery of sequence analysis programs is run on input genomic sequence. The purpose of these computes is to identify and Mask repeats and to assemble protein EST and mRNA alignments that will be used to inform MAKER’s gene-annotation process, which is outlined in steps 4 and 5 below. The default MAKER configuration uses four external programs: RepeatMasker (http://repeatmasker.org), BLAST (Altschul et al. 1990; Korf et al. 2003), Exonerate (Slater and Birney 2005), and SNAP (Korf 2004). Each is publicly available and free for academic use. All four programs are also easy to install and run on UNIX, Linux, and OS X.
Unless repeats are effectively masked, gene predictions and gene annotations will contain portions of transposons and viruses. MAKER uses a two-tier process to avoid this problem. First, RepeatMasker is used to screen the genome for low-complexity repeats; these are then “soft-masked,” e.g., transformed to lowercase letters rather than to Ns. Soft masking excludes these regions from nucleating BLAST alignments (Korf et al. 2003) but leaves them available for inclusion in annotations, as many protein-coding genes contain runs of low complexity sequence. MAKER also uses BLASTX together with an internal library of transposon and virally encoding proteins to identify mobile-elements. This process has been shown to substantially improve repeat masking as it identifies genome regions that are distantly related to the protein coding portions of transposons and viruses; these tend to be missed by RepeatMasker’s nucleotide-based alignment process, even when genome specific repeat libraries are available (Smith et al. 2007). Repeat regions identified in this process are masked to Ns. MAKER performs all of the actions automatically.
BLAST is used throughout the compute phase, first for repeat identification with RepeatMasker (as described above) and then to identify EST, mRNAs, and proteins with significant similarity to the input genomic sequence. Because BLAST does not take splice sites into account, its alignments are only rough approximations. MAKER therefore uses Exonerate (Slater and Birney 2005), a splice-site aware alignment algorithm to realign, or polish, sequences following filtering and clustering (see steps 2 and 3, below). Exonerate’s ability to align both protein and nucleotide sequences to the genome make it an economical choice for this task.
Filtering consists of identifying and removing marginal predictions and sequence alignments on the basis of scores, percent identities, etc. Filtering criteria for each external executable are set by modifying the text-based maker_bopts.ctl file (see configuration README distributed with MAKER). New users are not expected to edit this file, but advanced users may do so to change the behavior of the program. After filtering, the remaining data are then clustered against the genomic sequence to identify overlapping alignments and predictions. Clustering has two purposes. First, it groups diverse computational results into a single cluster of data, all of which support the same gene or transcript. Second, it identifies redundant evidence. For example, highly expressed genes may be supported by hundreds if not thousands of identical ESTs. Clustering criteria are set in the maker_bopts.ctl file, which instructs MAKER to keep some maximum number of members within each cluster, sorted on some series of filtering attributes such as score or fraction of the hit-sequence aligned. The default parameters are appropriate for most applications but can be easily modified.
This step realigns BLAST hits using a second alignment algorithm to obtain greater precision at exon boundaries. MAKER uses Exonerate (Slater and Birney 2005) to realign matching and highly similar ESTs, mRNAs, and proteins to the genomic input sequence. Because Exonerate takes splice-sites into account when generating its alignments, they provide MAKER with information about splice donors and acceptors. This information is especially useful in the synthesis and annotation steps (see below). The thresholds in the maker_bopts.ctl file earmark BLAST hits for polishing and are suitable for most applications but can be easily altered if desired (see configuration README distributed with MAKER).
MAKER synthesizes information from the polished and clustered EST and protein alignments to produce evidence for annotations. To do so, it identifies ESTs that it judges correspond to the same alternatively splice transcript. This is accomplished by comparing the coordinates of each polished sequence alignment on the genomic sequence in the same way that a human annotator might, e.g., by looking for internal exons with differing boundaries. Next MAKER identifies those protein alignments whose coordinates are consistent with each of the EST splice forms. Once a set of EST and protein alignments—all consistent with the same spliced transcript—has been identified, positions on the genomic input sequence upstream and downstream of the alignments are labeled as possible intergenic regions. Those bases on the genomic sequence that fall between exons are labeled as putative introns, and bases overlapping the protein alignments are labeled as putative translated sequence. MAKER then calculates a score for each of these nucleotides on the query sequence based upon the percentage of similarity of the alignment, type of alignment, and a query nucleotide’s position within the alignment. These scores together with their putative sequence types, e.g., Intergenic, Coding, Intron, and UTR, are then passed to SNAP. Based upon this information, SNAP then modifies its internal Hidden Markov Model (HMM). In the absence of any supporting EST or protein alignments, MAKER uses SNAP’s ab initio prediction (for additional details, see Training SNAP).
MAKER also post-processes the synthesis-generated SNAP predictions and recombines them with evidence to generate complete annotations. Each synthesis-generated SNAP prediction is checked against all ESTs and mRNAs, and 5′ and 3′ UTRs consistent with the prediction are identified based upon their coordinates relative to the predicted coding exons. The coordinates of the SNAP prediction are then altered to include these regions. This process is repeated for each of the synthesis-based predictions. Finally, compute evidence supporting each exon is added, and alternatively spliced forms are documented.
Additional details regarding MAKER’s architecture and implementation can be found in the release materials. All MAKER source code is publicly available; the current release along with installation instructions and documentation is available at http://www.yandell-lab.org/maker.
The input to MAKER is a genomic sequence (of any length) in fasta format and three configuration files describing external executable, sequence database locations, and various compute parameters (see configuration README distributed with MAKER). MAKER also uses four sequence database files during the compute phase: a transposons file, an optional repeatmasker database file, a proteins file, and anESTs/mRNAs file. Each file is in fasta format. The transposons file is bundled with MAKER and contains a selection of known transposon and virally encoded protein sequences. This file is used to identify and mask repeats missed by RepeatMasker, as this has been shown to substantially improve accuracy (Smith et al. 2007). In cases where no organism-specific repeat library is available, MAKER will automatically use thetransposon file to mask mobile elements and the RepeatMasker program to identify and mask low-complexity sequences. The repeatmasker file is an optional fasta file containing organism specific repeat sequences, if available. The proteins file contains any proteins users would like aligned to the genome. We recommend they use the latest version of the SWISS-PROT database for this purpose (Bairoch and Apweiler 2000). Finally, users should also supply a file of ESTs and/or mRNAs sequences derived from the organism being annotated. Assembling these into contigs is helpful, but it is not required.
MAKER outputs GMOD-compliant annotations in GFF3 format (http://www.sequenceontology.org/gff3.shtml) containing alternatively spliced transcripts, UTRs, and evidence for each gene’s annotated transcript and protein sequences. This file can be directly imported into genome browsers and databases that adhere to Sequence Ontology (Eilbeck et al. 2005) and GMOD (http://www.gmod.org) standards. For convenience, MAKER also outputs multifasta files of transcripts and protein sequences for both annotations and ab initio SNAP predictions.
MAKER also writes a GAME XML file (http://www.fruitfly.org/annot/apollo/game.rng.txt) containing the same contents as the corresponding GFF3 file (http://www.sequenceontology.org/gff3.shtml); this file can be directly viewed in the Apollo genome browser (Figure 3) (Lewis et al. 2002). Apollo can also be used to directly edit annotations and to save them to GFF3 format, thus changes to MAKER annotations can be saved prior to uploading them into a GMOD browser or database. Apollo can also directly export the revised transcripts and protein sequences in fasta format. This is an especially useful feature for those seeking to annotate a single contig or BAC rather than an entire genome, as it circumvents the overhead associated with creating and maintaining a GMOD database. Figure 3 shows a portion of an annotated contig viewed in Apollo genome browser. Compute evidence assembled by MAKER is shown in the top panel; its resulting annotation, below. This figure demonstrates how MAKER synthesizes data gathered by its compute pipeline into evidence-informed gene annotations; while SNAP produced two ab initio predictions in this region, the EST and protein alignments clearly support a single gene. Note too the 3′ UTR on the MAKER annotation derived from the EST alignments.
Compute data are essential for discriminating real genes from false positives. To simplify the quality evaluation process, each MAKER-annotated transcript has an associated quality index included in its GFF3 and GAME XML outputs. This is a nine-dimensional summary (Table 2) of a transcript’s key features and how they are supported by the data gathered by MAKER’s compute pipeline. The quality index associated with the mRNA shown in Figure 3 is QI:0|0.77|0.68|1|0.77|0.78|19|462|824.
Quality indices play a central role in training MAKER for a particular genome, where they are used to identify transcripts that are well supported by EST and protein evidence but poorly supported by ab initio SNAP predictions. These cases are used to retrain SNAP via the bootstrap procedure outlined below. MAKER’s quality indices also provide an easy means to sort and rank transcripts by key features such as number of exons, presence or absence of UTR, or degree of computational support. Quality indices were used to assemble the HC S. mediterranea genes described in the Results section.
For optimal accuracy, a gene finder must be trained for a specific genome (Korf 2004), generally using several hundred existing gene-annotations drawn from a body of experimental data gathered over many years. Unfortunately, many emerging genomes do not have a history of experimental molecular biology. It has therefore become a common practice to use gene finders trained in one genome to predict genes in another—a far from optimal solution to the problem (for discussion, see Korf 2004). Information gathered from ab initio predictions is essential for the annotation process, even when other evidence is available. Moreover, in the absence of experimental evidence and sequence similarities, the probabilistic models produced by ab initio gene prediction programs offer the best guesses at gene structure. The SNAP (Korf 2004) gene finder was designed from the outset to be easily configured for any genome; hence its use in MAKER.
MAKER is trained for a genome using a two-step process. First, SNAP is trained by aligning a set of universal genes to the input genome (Parra et al. 2007). These universal genes are highly conserved in all eukaryotes and can be identified using pairwise and profile-HMM alignment methods. The resulting gene structures are used to create a first-pass version of SNAP for use in the next stage of the training process. This initial stage of the training procedure is automated, and complete details of the process can be found in the MAKER README. More extensive documentation is provided by Parra et al. (2007).
The genome-specific HMM produced in the first stage of SNAP training is further refined with a second stage of training. This is accomplished by running MAKER on a few megabases of genomic sequence (enough to result in a few hundred annotations). The resulting GFF3 outputs are then used as inputs to a script called maker2zff.pl, whose output is a ZFF file that can be used to automatically create a revised HMM. The maker2zff.pl script uses the quality index MAKER attaches to each annotation to identify a set of gene models with intron-exon structures that are unambiguously supported by EST alignments and protein homology. These genes are then used to further refine the SNAP HMM. The maker2fzff.pl script is bundled with MAKER, and programs necessary to create the HMM are included in the SNAP package. To train MAKER for the S. mediterranea genome, we first trained SNAP using the universal gene set as outlined above. We then ran MAKER on a randomly selected 100-Mb portion of the S. mediterranea genome (∼10% of the entire genome). The resulting GFF3 files were used as inputs to maker2zff.pl, and the refined SNAP-HMM was used in the final annotation run.
MAKER is available for download from http://www.yandell-lab.org/downloads/maker/maker.tar.gz. Once downloaded, the MAKER package should be unzipped and untared. Full installation and usage instructions are available in the file called README.
The GFF3 output files generated by MAKER were used to populate SmedGD. The files were uploaded into a mySQL database, using a standard Bioperl (http://www.bioperl.org) loading script, bp_seqfeature_load.pl. This script converts GFF3 formatted annotations to Bio∷SeqFeatureI objects, which are stored in the mySQL database. GBrowse, a tool distributed by GMOD (http://www.gmod.org) implementing a Bio∷DB∷SeqFeature∷Store database adaptor, accesses and displays rows of data or tracks that are mapped to specific locations in the genome. SmedGD consists of MAKER annotations as well as project specific features, such as additional protein homology, human curated genes, and RNA interference phenotype data. The database is available at http://smedgd.neuro.utah.edu.
实际使用:
待添加。。。
freemao
FAFU
标签:
原文地址:http://www.cnblogs.com/freemao/p/4850593.html