标签:
软件下载与说明:http://www.broadinstitute.org/software/allpaths-lg/blog/?page_id=12
原始数据的深度要达到100以上。
至少要两个库,大库和小库,
小库的一对reads要有重叠部分。并且小库的插入片段大小分布差异要在20%以内。
大库插入片段要接近3000,并且长度分布可以有较大的差异。
ALLPATHS‐LG requires a minimum of 2 paired‐end libraries – one short and one long. The short library average separation size must be slightly less than twice the read size, such that the reads from a pair will likely overlap – for example, for 100 base reads the insert size should be 180 bases. The distribution of sizes should be as small as possible, with a standard deviation of less than 20%. The long library insert size should be approximately 3000 bases long and can have a larger size distribution. Additional optional longer insert libraries can be used to help disambiguate larger repeat structures and may be generated at lower coverage.
A fragment library is a library with a short insert separation, less than twice the read length, so that the reads may overlap (e.g., 100bp Illumina reads taken from 180bp inserts.) A jumping library has a longer separation, typically in the 3kbp‐10kbp range, and may include sheared or EcoP15I libraries or other jumping‐library construction; ALLPATHS can handle read chimerism in jumping libraries. Note that fragment reads should be long enough to ensure the overlap.
A fragment library is a library with a short insert separation, less than twice the read length, so that the reads may overlap (e.g., 100bp Illumina reads taken from 180bp inserts.) A jumping library has a longer separation, typically in the 3kbp‐10kbp range, and may include sheared or EcoP15I libraries or other jumping‐library construction; ALLPATHS can handle read chimerism in jumping libraries. Note that fragment reads should be long enough to ensure the overlap.
现在也可以加入pacbio数据,但是只针对真菌基因组。
如果你有reference genome就提供

allpaths需要的输入文件:
1, DATA directory里的base,quality score, and pairing information files.like that:
<REF>/<DATA>/frag_reads_orig.fastb
<REF>/<DATA>/frag_reads_orig.qualb
<REF>/<DATA>/frag_reads_orig.pairs
<REF>/<DATA>/jump_reads_orig.fastb
<REF>/<DATA>/jump_reads_orig.qualb
<REF>/<DATA>/jump_reads_orig.pairs
2, a ploidy file must also be present.the file polidy file is a single-line file containing a number.The specfic file name is :
<REF>/<DATA>/ploidy
如何产生这些输入文件呢:
用自带的perl脚本: PrepareALLPaths.pl。用这个脚本需要提供两个配置文件:
in_groups.csv和 in_libs.csv。
csv的意思是: comma-separated-values
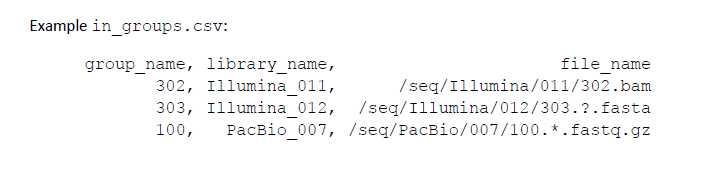
首先来看in_groups.csv文件:
group_name: a UNIQUE nickname for this specific data set.
library_name: the library to which the data set belongs.
file_name: the absolute path to the data file.
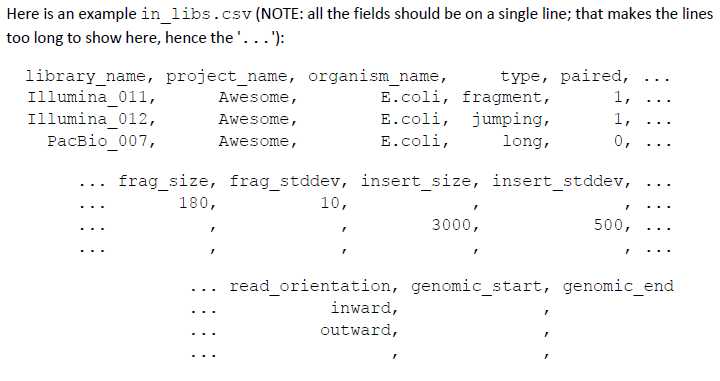
再看in_libs.csv文件:这个文件是描述你的library的。
library_name: matches the same field in in_groups.csv.
project_name: a string naming the project.
organism_name: the organism.
type: fragment, jumping, EcoP15, etc. This field is only informative.
paired: 0: Unpaired reads; 1: paired reads.
frag_size: average number of bases in the fragments (only defined for FRAGMENT libraries).
frag_stddev: estimated standard deviation of the fragments sizes (only defined for FRAGMENT libraries).
insert_size: average number of bases in the inserts (only defined for JUMPING libraries; if larger than 20 kb, the library is considered to be a LONG JUMPING library).
insert_stddev: estimated standard deviation of the inserts sizes (only defined for JUMPING libraries).
read_orientation: inward or outward. Outward oriented reads will be reversed.
genomic_start: index of the FIRST genomic base in the reads. If non‐zero, all the bases before genomic_start will be trimmed out.
genomic_end: index of the LAST genomic base in the reads. If non‐zero, all the bases after genomic_end will be trimmed out.
这两个文件准备好以后,就可以run 这个perl脚本了
PrepareALLPathsInputs.pl \
DATA_DIR=‘full path to REFERENCE DIR‘/mydata \
PICARD_TOOLS_DIR=‘path to picard tools‘ \
IN_GROUPS_CSV=in_groups.csv \
IN_LIBS_CSV=in_libs.csv \
INCLUDE_NON_PF_READS=0 \
PHRED_64 = 0 \
PLOIDY = 2 \
DATA_DIR: is the location of the ALLPATHS DATA directory where the converted reads will be placed.
PICARD_TOOLS_DIR: is the path to the Picard tools needed for data conversion, if your data is in BAM format.
IN_GROUPS_CSV: 编辑的in_groups.csv的文件位置,如果在当前目录,可以不写。
IN_LIBS_CSV: 编辑的in_libs.csv的文件位置,如果在当前目录,可以不写。
INCLUDE_NON_PF_READS: 0代表只有paired end reads, 1表示含有non_PF reads
PHRED_64: 0表示碱基质量是按照phred_33,1表示碱基质量按照phred_64
PLOIDY: 产生polidy文件,是单倍体就是1,二倍体是2.
这个脚本执行完后,ALLPATHS所需的输入文件就都准备好了~
下一步,运行ALLPATHS,
RunAllPathsLG
PRE=<user pre>
DATA_SUBDIR=mydata
RUN=myrun
REFERENCE_NAME=staph
TARGETS=standard
This will create (if it doesn’t already exist) the following pipeline directory structure:
<user pre>/staph/mydata/myrun
Where staph is the REFERENCE directory, mydata is the DATA directory containing the imported data, and myrun is the RUN directory.
实际使用时的命令:
freemao
FAFU
标签:
原文地址:http://www.cnblogs.com/freemao/p/4885039.html