标签:
针对组合 4 - 10 - 14 - 18
分别对正交4,反交10, 亲本14和18进行DNA测序
对正反交子代的根和叶三个重复进行RNA测序
4的DNA数据,测序深度28。
/share/fastq/data/Unaligned_016_05_31-a/Project_offspring-DNA/Sample_4/4_TGACCA_L000_R1.fastq.gz
/share/fastq/data/Unaligned_016_05_31-a/Project_offspring-DNA/Sample_4/4_TGACCA_L000_R2.fastq.gz
10的DNA数据,测序深度31。
/share/fastq/data/Unaligned_016_05_31-a/Project_offspring-DNA/Sample_10/10_TAGCTT_L000_R1.fastq.gz
/share/fastq/data/Unaligned_016_05_31-a/Project_offspring-DNA/Sample_10/10_TAGCTT_L000_R2.fastq.gz
以及L4-1, L4-2, L4-3, R4-1, R4-2, R4-3, L10-1, L10-2, L10-3, R10-1, R10-2, R10-3
对原始数据进行质量控制。
用fastqc查看测序质量
/share/bioinfo/miaochenyong/Myscripts/preprocess_raw_data/fastqc -t 20 *gz
对低质量reads用trimmomatic进行过滤
python /share/bioinfo/miaochenyong/Myscripts/preprocess_raw_data/TrimOnCluster.py fq1 fq2 headcrop_n(需要crop 前面碱基的)
python /share/bioinfo/miaochenyong/Myscripts/preprocess_raw_data/TrimOnCluster.py fq1 fq2 (普适,不进行crop,只进行简单的质量过滤)
过滤都会去除接头序列,中心测序接头文件在/share/bioinfo/miaochenyong/Myscripts/preprocess_raw_data/adapters.fa
过滤完之后,会生成对应的paired和unpaired文件。下面的分析只用paired文件。需要改名的话有个脚本将paired去掉:
/share/bioinfo/miaochenyong/Myscripts/preprocess_raw_data/rename.py 再手动修改后续脚本接受的名称。
过滤完后可以再运行fastqc,看下过滤后的质量怎么样。
所有过滤后的数据都已经转移至移动4T移动硬盘。
接下来对亲本14和18进行基因组重构。
建立各自的重构目录,将各自的DNA数据链接到该目录下
先用bowtie2比对然后call SNP
python /share/bioinfo/miaochenyong/Myscripts/call_snp_pipeline/bowtie2OnCluster.py /share/bioinfo/miaochenyong/ASE-PROJECT/Osativa_323_v7.0.fa
筛选只有1/1的SNP
python /share/bioinfo/miaochenyong/Myscripts/vcf_filter/parents_filter-DNA.py > onlyALTSNP.vcf
创建子目录,利用SNP和原始参考基因组,生成新的基因组。
java -jar /share/bioinfo/miaochenyong/ASE-PROJECT/GenomeAnalysisTK.jar -T FastaAlternateReferenceMaker -R old.fa -o new.fa -V onlyALTSNP.vcf
用vim修改新基因组中染色体的名字,使得和注释文件保持一致。
vim new.fa
对新基因组创建比对index, dict, fai
bowtie2-build P1round1.fa P1round1
java -jar /share/Public/cmiao/picard-tools-1.112/CreateSequenceDictionary.jar R=P2round3.fa O=P2round3.dict
samtools faidx P2round3.fa
进行多伦构建,直到所call snp 数目趋于稳定为止。
目录结构如下图所示:
两个参考基因组都创建好后,用/share/bioinfo/miaochenyong/Myscripts/call_snp_pipeline/bowtie2OnCluster.py将4和10分别对比到重构的亲本P1和P2.
用/share/bioinfo/miaochenyong/Myscripts/call_snp_pipeline/tophatOnCluster.py将RNA数据比对到P1和P2.
DNA处理步骤如下:
1, bowtie2比对+sam转成bam
2,sort bam
3, rmdup
4, index 之后就可以call SNP
RNA数据处理如下:
1, tophat2比对
2, 链接accepted_hits.bam到新名字
3, rmdup
4, read group
5, index
6,splitN
7,index 之后就可以call SNP
DNA和RNA数据都处理完后,创建如下目录:
将比对到P1的最终结果全部链接到P1_All_Bams, 将比对到P2的最终结果全部链接到P2_All_Bams。如图所示:
然后在两个目录下,分别执行:
python python /share/bioinfo/miaochenyong/Myscripts/call_snp_pipeline/SNPonCluster.py /share/workplace/home/cmiao/ASE/16-personal-genome/new-genome7/P2round7.fa
脚本会对DNA和RNA合并在一起CALL SNP。
结束后,会在各自目录生成SNP 结果文件FB.vcf
分别在两个目录运行:
1, 过滤i.ALT==1, P1GT和P2GT不能含有0/1且不能相同(可能一个是None,即有一个没有比对上)
nohup python ~/ASE-final-tackle/step1-filter_GT-p1p2DNA.py FB.vcf step1.P1.vcf 14 18 &
nohup python ~/ASE-final-tackle/step1-filter_GT-p1p2DNA.py FB.vcf step1.P2.vcf 14 18 &
2, MNP 转化为SNP
nohup python ~/ASE-final-tackle/step2-filter-continuous-snp.py step1.P1.vcf > step2.P1.vcf &
nohup python ~/ASE-final-tackle/step2-filter-continuous-snp.py step1.P2.vcf > step2.P2.vcf &
以上两步时间较长,放到后台运行。
将得到的两个文件链接到Analysis目录下
3-1,得到两个文件共同位点 step1-filter_GT-p1p2DNA.py
3-2, 对共同位点筛选 step3-2-common_POSandBASE.py
4, 将各自vcf转化为AO RO文件 step4-vcf2AORO.py
5, 将两个AO RO文件合并为一个,原则是,两个数字比较取较大者。step5-combine-table.py
6, 由于文件中同时含有子代DNA, 子代RNA叶和根,因为在非基因区,RNA数据可能没有信息,或者基因有的不表达,也是没有信息的,因此将这些行去掉 step6-rmdot.py
7, 对上步文件进行二项分布检验。step7-binomial.py
8, 加上gene信息。step8-addGene.py
以上脚本均在~/ASE-final-tackle下。
为了加快速度,对vcf文件进行分割
python ~/ASE-final-tackle/splitvcf.py P1tmp 100000
python ~/ASE-final-tackle/splitvcf.py P2tmp 100000
然后一起执行 3-2到8步骤:
python ~/ASE-final-tackle/batchASEstep2-8.py 13 15 7,1-plus,L7-1,L7-2,L7-3,L1-1,L1-2,L1-3,R7-1,R7-2,R7-3,R1-1,R1-2,R1-3 9
执行完后进行合并文件,将想合并的文件合并:
python ~/ASE-final-tackle/combineSplited.py step3.P1 step3.P1.vcf
python ~/ASE-final-tackle/combineSplited.py step3.P2 step3.P2.vcf
python ~/ASE-final-tackle/combineSplited.py step5 step5.combined.map
python ~/ASE-final-tackle/combineSplited.py step6 step6.rmpDOT.map
python ~/ASE-final-tackle/combineSplited.py step7 step7.stated.map
python ~/ASE-final-tackle/combineSplited.py step8 step8.addGene.map
对结果进行简单统计,并生成各个样品对应的ASE文件:
python ~/ASE-final-tackle/stat_ASE_types.py step8.addGene.map > stats.log
对结果进行统计:
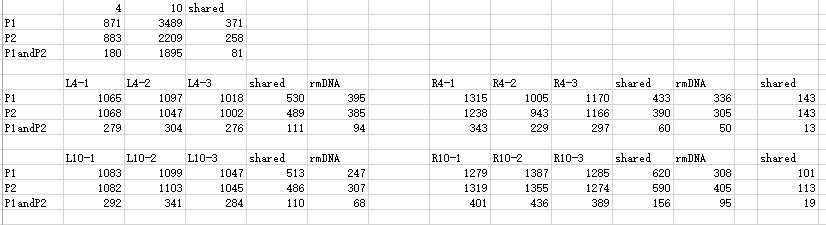
用到两个脚本:
python ~/ASE-final-tackle/getCommonGenes.py file1,file2 out (对两个文件取交集)
python ~/ASE-final-tackle/getCommonGenes.py file1,file2,file3 out (对三个文件取交集)
python ~/ASE-final-tackle/MinusDNA.py R2P1.txt 8_P1_ASEgenes.txt R2p1_rmDNA.txt(将RNA中ASE基因减去同样在DNA中表现出ASE的基因)
对感兴趣的ASE基因进行GO分析,将ASE基因复制到http://rice.plantbiology.msu.edu/downloads_gad.shtml 选择GOSlim Assignments 然后提交
将得到的文件复制到txt, 转移至服务器,执行:
python ~/ASE-final-tackle/extractGO.py L39P2GOraw.txt > L39P2GO.txt 得到基因对应的GO号。
将该文件上传至http://wego.genomics.org.cn/cgi-bin/wego/index.pl 选择WEGO Native Format上传。
选择level 3, 看summary结果,。然后plot,基因数量不用log显示,生成图片查看富集情况。
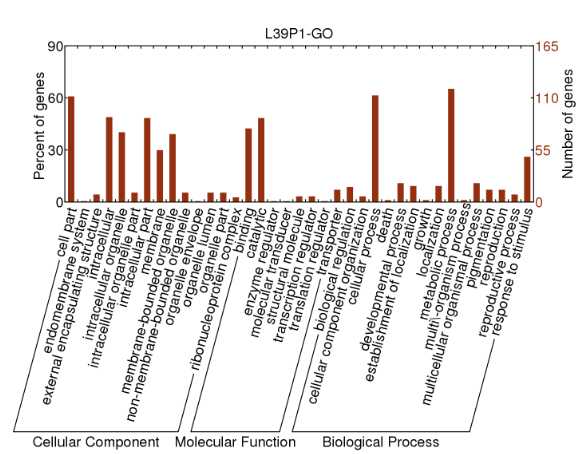
对ASE类型进行统计:
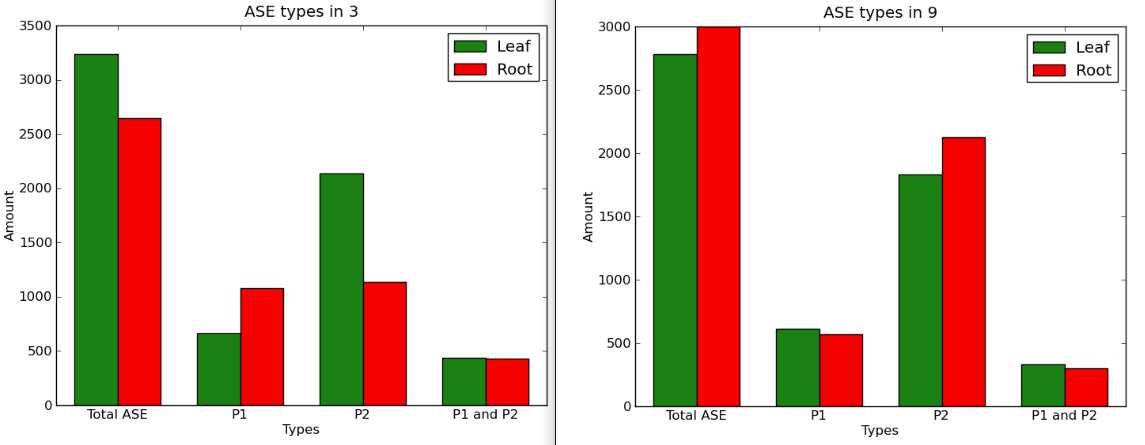
每个项目用wc -l 进行统计
画图的配置文件:/share/workplace/home/cmiao/MCY-ASE-Final/matplotlibrc 改为AGG
画图脚本:python /share/workplace/home/cmiao/MCY-ASE-Final/drawbar1.py L-ASE-List R-ASE-List F1name
对无论正反交都不变的特殊ASE进行统计画图
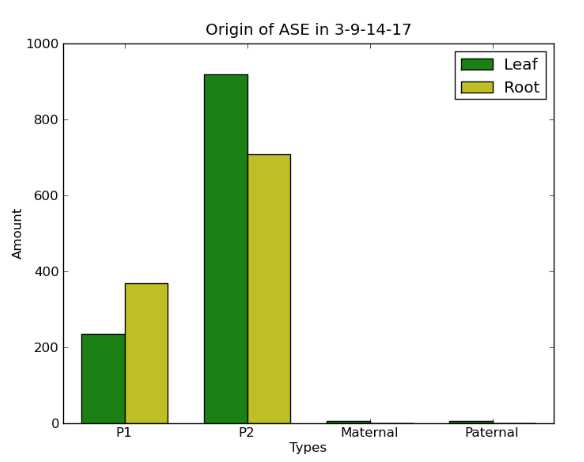
统计了无论正反交来自于P1, P2, 母本,父本
用~/ASE-final-tackle/getCommonGenes.py 和 wc -l 统计
画图脚本:
python ~/ASE-final-tackle/ASE-origin.py L-specialASE-List R-specialASE-List F1name
在ASE位点处,分析12中SNP组合的分布,看是否由于测序影响导致的。
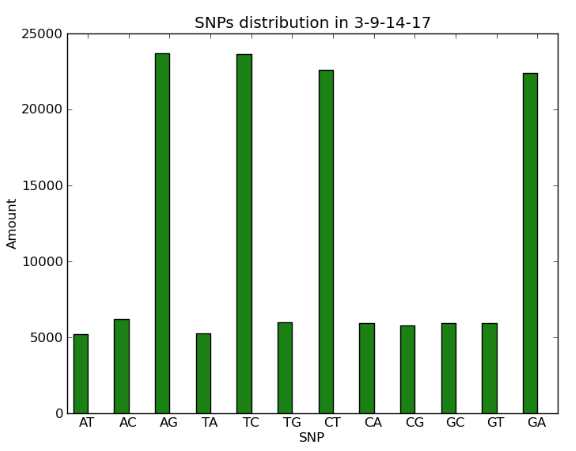
得到各类SNP数量: python ~/ASE-final-tackle/classifySNP.py step6file step7file
画图: python ~/MCY-ASE-Final/drawbarSNP.py SNPlist F1name
看ASE在各个染色体的分布:
python ~/ASE-final-tackle/classifyChr.py step8file
标签:
原文地址:http://www.cnblogs.com/freemao/p/5672225.html