标签:strong 可视化 range san libc visio dna from orm
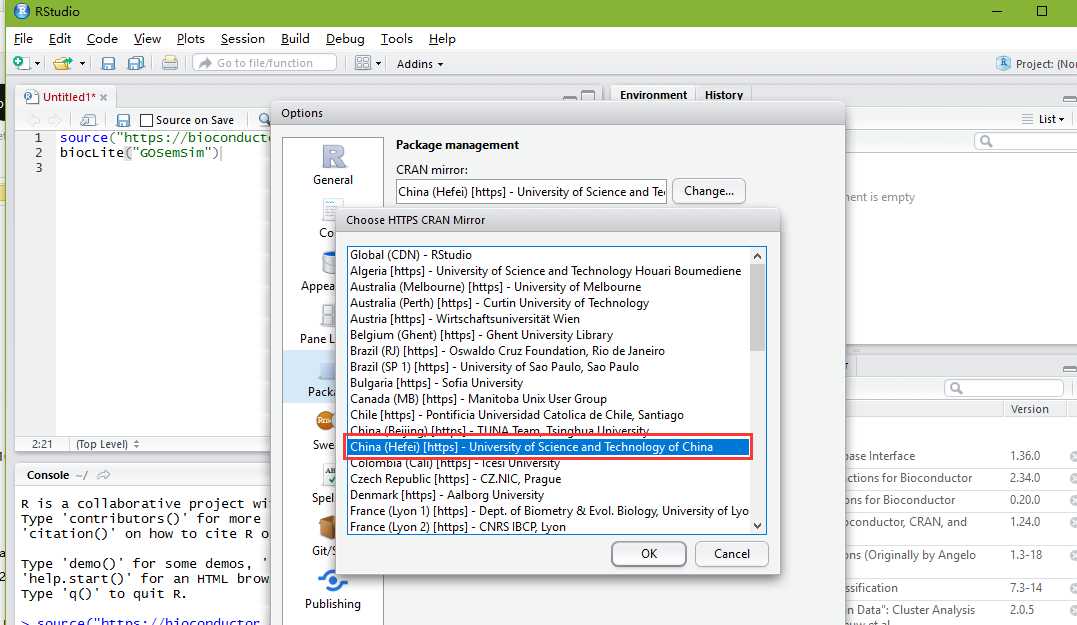
Install GOSemSim is easy, follow the guide in the Bioconductor page:
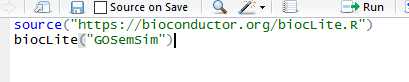
## try http:// if https:// URLs are not supportedsource("https://bioconductor.org/biocLite.R")## biocLite("BiocUpgrade") ## you may need thisbiocLite("GOSemSim")


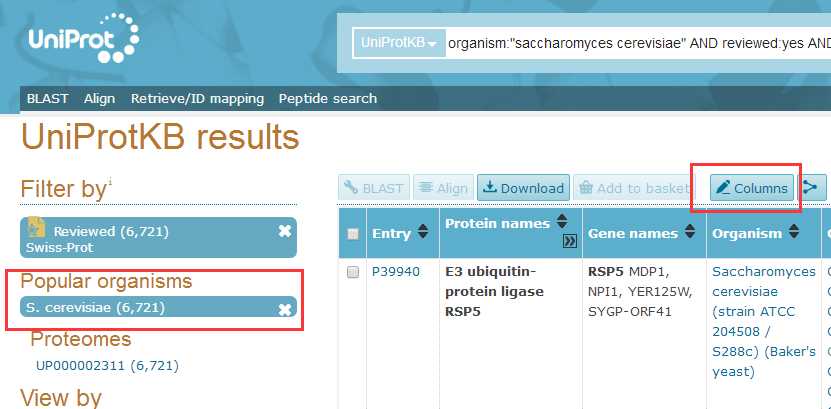

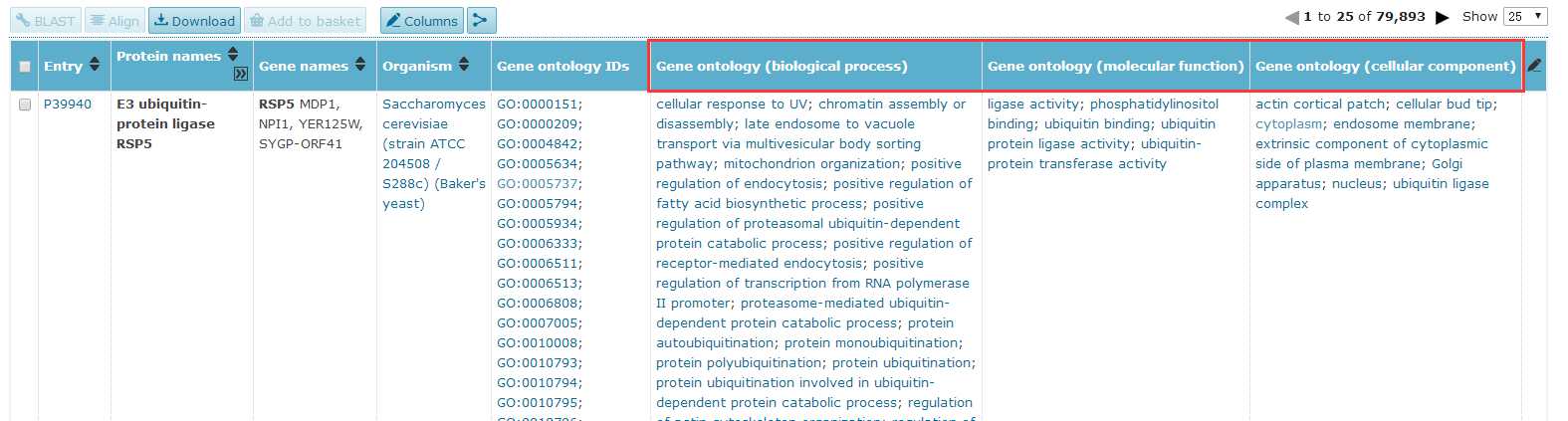
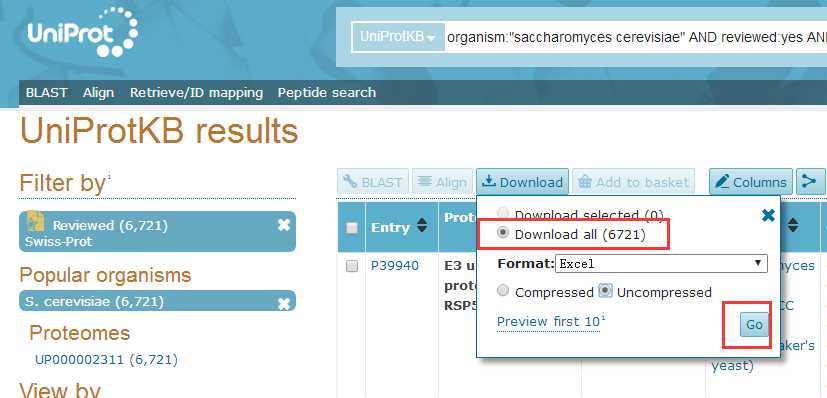
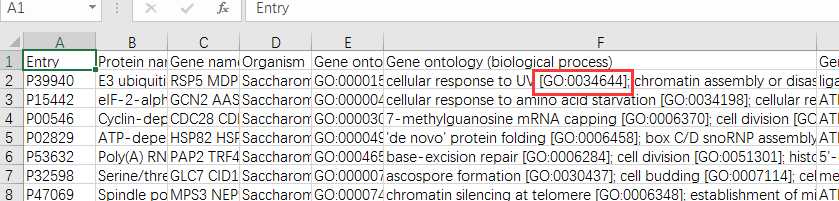
# -*- coding: utf-8 -*-"""Created on Fri Oct 28 19:04:38 2016@author: sun"""import pandas as pdimport reyeast=pd.read_csv(‘yeast.csv‘)#Gene ontology (biological process)#Gene ontology (molecular function)#Gene ontology (cellular component)bp=yeast[‘Gene ontology (biological process)‘]bp=bp.fillna(value=‘‘)for i in range(len(bp)): temp=re.findall(r"GO:\d{7}",bp[i]) bp[i]=‘;‘.join(temp)mf=yeast[‘Gene ontology (molecular function)‘]mf=mf.fillna(value=‘‘)for i in range(len(mf)): temp=re.findall(r"GO:\d{7}",mf[i]) mf[i]=‘;‘.join(temp)cc=yeast[‘Gene ontology (cellular component)‘]cc=cc.fillna(value=‘‘)for i in range(len(cc)): temp=re.findall(r"GO:\d{7}",cc[i]) cc[i]=‘;‘.join(temp)yeast[‘Gene ontology (biological process)‘]=bp yeast[‘Gene ontology (molecular function)‘]=mf yeast[‘Gene ontology (cellular component)‘]=cc yeast.to_csv(‘go.csv‘,index=False,columns =[‘Entry‘,‘Gene ontology (cellular component)‘,‘Gene ontology (molecular function)‘,‘Gene ontology (biological process)‘])

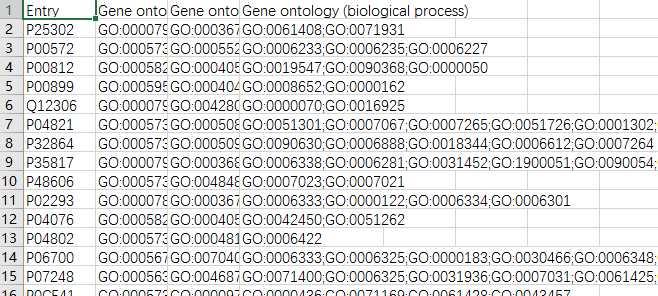
yeast=pd.read_csv(‘yeast_gold_protein_pair.csv‘)go=pd.read_csv(‘go.csv‘,index_col=0)protein_a=go.loc[yeast.idA,:]protein_b=go.loc[yeast.idB,:]protein_a.to_csv(‘GOProteinA.csv‘)protein_b.to_csv(‘GOProteinB.csv‘)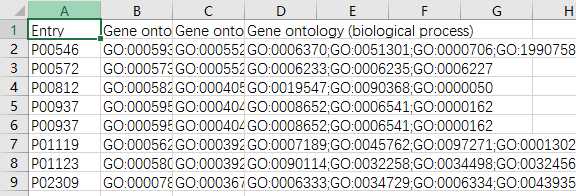
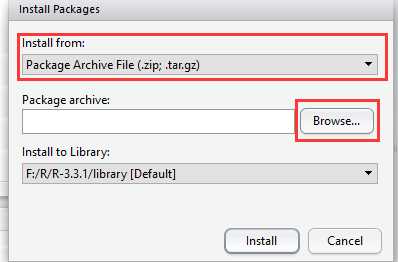
 没什么好说的,直接双击安装即可。注意:不能装到带有空格的目录中
没什么好说的,直接双击安装即可。注意:不能装到带有空格的目录中
 也没什么好说,双击直接安装就行了。
也没什么好说,双击直接安装就行了。

For IC-based methods, information of GO term is species specific. We need to calculate IC for all GO terms of a species before we measure semantic similarity. GOSemSim support all organisms that have an OrgDb object available.
Bioconductor have already provided OrgDb for about 20 species, seehttp://bioconductor.org/packages/release/BiocViews.html#___OrgDb.
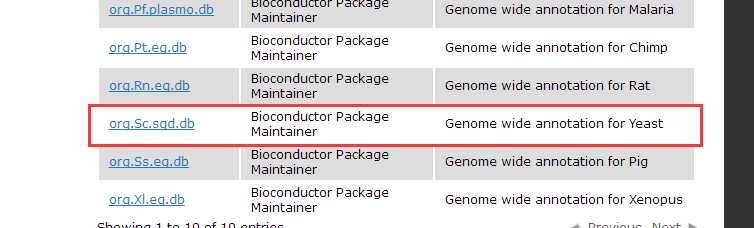
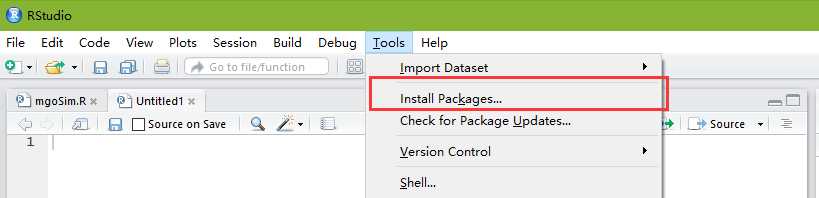
Once we have OrgDb, we can build annotation data needed by GOSemSim via godata function.
library(GOSemSim)hsGO <- godata(‘org.Hs.eg.db‘, ont="MF")## [1] "preparing gene to GO mapping data..."
## [1] "preparing IC data..."User can set computeIC=FALSE if they only want to use Wang’s method.
In GOSemSim, we implemented all these IC-based and graph-based methods. goSim function calculates semantic similarity between two GO terms, while mgoSim function calculates semantic similarity between two sets of GO terms.
goSim("GO:0004022", "GO:0005515", semData=hsGO, measure="Jiang")## [1] 0.155goSim("GO:0004022", "GO:0005515", semData=hsGO, measure="Wang")## [1] 0.158go1 = c("GO:0004022","GO:0004024","GO:0004174")go2 = c("GO:0009055","GO:0005515")mgoSim(go1, go2, semData=hsGO, measure="Wang", combine=NULL)## GO:0009055 GO:0005515
## GO:0004022 0.205 0.158
## GO:0004024 0.185 0.141
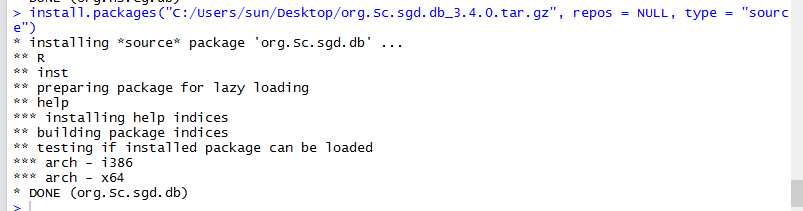
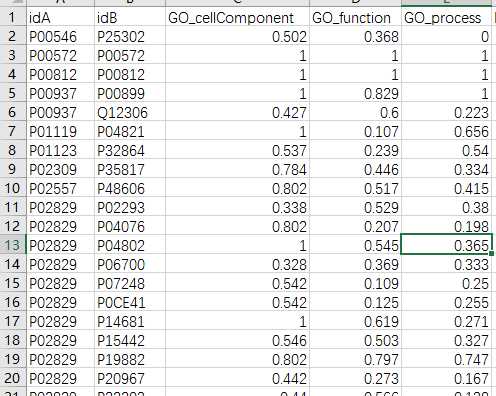
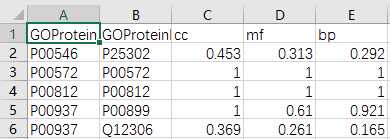
## GO:0004174 0.205 0.158mgoSim(go1, go2, semData=hsGO, measure="Wang", combine="BMA")## [1] 0.192> library(GOSemSim)> hsGO <- godata(‘org.Hs.eg.db‘, ont="MF")[1]"preparing gene to GO mapping data..."[1]"preparing IC data..."> goSim("GO:0004022","GO:0005515", semData=hsGO, measure="Jiang")[1]0.155> goSim("GO:0004022","GO:0005515", semData=hsGO, measure="Wang")[1]0.158> go1 = c("GO:0004022","GO:0004024","GO:0004174")> go2 = c("GO:0009055","GO:0005515")> mgoSim(go1, go2, semData=hsGO, measure="Wang", combine=NULL) GO:0009055 GO:0005515GO:00040220.2050.158GO:00040240.1850.141GO:00041740.2050.158> mgoSim(go1, go2, semData=hsGO, measure="Wang", combine="BMA")[1]0.192library(GOSemSim)library(org.Sc.sgd.db)GOProteinA<- read.csv("GOProteinA.csv", stringsAsFactors = F)GOProteinB<- read.csv("GOProteinB.csv", stringsAsFactors = F)cc <- c()mf <- c()bp <- c()scGo <- godata(‘org.Sc.sgd.db‘, ont ="cc", computeIC = F)for(i in1:length(GOProteinA$Entry)){ go1 <- c(strsplit(GOProteinA[i,2], split =";")[[1]]) go2 <- c(strsplit(GOProteinB[i,2], split =";")[[1]]) cc[[i]]<- mgoSim(go1, go2, semData = scGo, measure ="Wang")}scGo <- godata(‘org.Sc.sgd.db‘, ont ="mf", computeIC = F)for(i in1:length(GOProteinA$Entry)){ go1 <- c(strsplit(GOProteinA[i,3], split =";")[[1]]) go2 <- c(strsplit(GOProteinB[i,3], split =";")[[1]]) mf[[i]]<- mgoSim(go1, go2, semData = scGo, measure ="Wang")}scGo <- godata(‘org.Sc.sgd.db‘, ont ="bp", computeIC = F)for(i in1:length(GOProteinA$Entry)){ go1 <- c(strsplit(GOProteinA[i,4], split =";")[[1]]) go2 <- c(strsplit(GOProteinB[i,4], split =";")[[1]]) bp[[i]]<- mgoSim(go1, go2, semData = scGo, measure ="Wang")}GOFeature<- data.frame(GOProteinA$Entry,GOProteinB$Entry, cc, mf, bp)write.csv(GOFeature,‘GOFeature.csv‘, na =‘0‘,#将nan值填充为0 row.names = FALSE)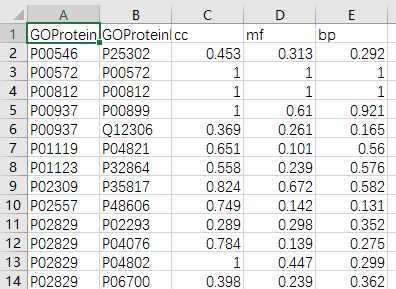
cellular bud neck [GO:0005935];cyclin-dependent protein kinase holoenzyme complex [GO:0000307];cytoplasm [GO:0005737];endoplasmic reticulum [GO:0005783];nucleus [GO:0005634]ATP binding [GO:0005524];cyclin-dependent protein serine/threonine kinase activity [GO:0004693];histone binding [GO:0042393];protein serine/threonine kinase activity [GO:0004674];RNA polymerase II core binding [GO:0000993]7-methylguanosine mRNA capping [GO:0006370];cell division [GO:0051301];meiotic DNA double-strand break processing [GO:0000706];mitotic sister chromatid biorientation [GO:1990758];negative regulation of double-strand break repair via nonhomologous end joining [GO:2001033];negative regulation of meiotic cell cycle [GO:0051447];negative regulation of mitotic cell cycle [GO:0045930];negative regulation of sister chromatid cohesion [GO:0045875];negative regulation of transcription, DNA-templated [GO:0045892];negative regulation of transcription from RNA polymerase II promoter during mitosis [GO:0007070];peptidyl-serine phosphorylation [GO:0018105];peptidyl-threonine phosphorylation [GO:0018107];phosphorylation of RNA polymerase II C-terminal domain [GO:0070816];positive regulation of meiotic cell cycle [GO:0051446];positive regulation of mitotic cell cycle [GO:0045931];positive regulation of nuclear cell cycle DNA replication [GO:0010571];positive regulation of spindle pole body separation [GO:0010696];positive regulation of transcription, DNA-templated [GO:0045893];positive regulation of transcription from RNA polymerase II promoter [GO:0045944];positive regulation of triglyceride catabolic process [GO:0010898];protein localization to nuclear periphery [GO:1990139];protein localization to nucleus [GO:0034504];protein phosphorylation involved in cellular protein catabolic process [GO:1902002];protein phosphorylation involved in DNA double-strand break processing [GO:1990802];protein phosphorylation involved in double-strand break repair via nonhomologous end joining [GO:1990804];protein phosphorylation involved in mitotic spindle assembly [GO:1990801];protein phosphorylation involved in protein localization to spindle microtubule [GO:1990803];regulation of budding cell apical bud growth [GO:0010568];regulation of double-strand break repair via homologous recombination [GO:0010569];regulation of filamentous growth [GO:0010570]; regulation of nucleosome density [GO:0060303];regulation of spindle assembly [GO:0090169]; regulation of telomere maintenance via telomerase [GO:0032210];synaptonemal complex assembly [GO:0007130]; vesicle-mediated transport [GO:0016192]nuclear chromatin [GO:0000790];SBF transcription complex [GO:0033309]DNA binding [GO:0003677];identical protein binding [GO:0042802];transcriptional activator activity, RNA polymerase II core promoter proximal region sequence-specific binding [GO:0001077]positive regulation of transcription from RNA polymerase II promoter in response to heat stress [GO:0061408];positive regulation of transcription involved in G1/S transition of mitotic cell cycle [GO:0071931]
ccA <- c(strsplit(GOProteinA[1,2], split =";")[[1]])ccB <- c(strsplit(GOProteinB[1,2], split =";")[[1]])mfA <- c(strsplit(GOProteinA[1,3], split =";")[[1]])mfB <- c(strsplit(GOProteinB[1,3], split =";")[[1]])bpA <- c(strsplit(GOProteinA[1,4], split =";")[[1]])bpB <- c(strsplit(GOProteinB[1,4], split =";")[[1]])scGo <- godata(‘org.Sc.sgd.db‘, ont ="cc")cc <- mgoSim(ccA, ccB, semData = scGo, measure ="Wang")scGo <- godata(‘org.Sc.sgd.db‘, ont ="mf")mf <- mgoSim(mfA, mfB, semData = scGo, measure ="Wang")scGo <- godata(‘org.Sc.sgd.db‘, ont ="bp")bp <- mgoSim(bpA, bpB, semData = scGo, measure ="Wang")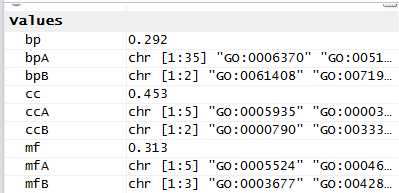

标签:strong 可视化 range san libc visio dna from orm
原文地址:http://www.cnblogs.com/ahusun/p/b9bd34877ae5b8092d3bd1857498b3f3.html