标签:工具 统计 http fusion uart oba 没有 mil 流程
原文出处:https://genomebiology.biomedcentral.com/articles/10.1186/s13059-016-0881-8
摘要:
RNA-seq已经被广泛的运用,但是现在还没有通行的分析流程。RNA-seq 分析的主要步骤包括:实验设计,QC控制,read alignment,quantification of gene and transcript leves, visualization, differential gene expression, alternative splicing, functional analysis, gene fusion detection and eQTL mapping。本文中我们重点强调每个步骤可能遇到的问题。
背景:
在基因组学研究中RNA-seq已经成为一种常规手段,是生命科学研究的一个标准研究工具。已有很多RNA-seq 分析流程已经建立。对于研究RNA-seq的新手来说,一个重要的挑战是如何在众多的分析工具中选择适合自己项目的。研究者会根据自己的研究目的和研究对象来制定合适的分析策略。比如,对于研究对象的基因组是已知的,把reads mapping到基因组上能验证转录子是否存在。相反的,如果研究对象的基因组是未知的,通过从头组装reads形成contigs,并将这些contigs mapping 到转录组上,能获得表达量的定量。对于有很好注释的基因组,如人类基因组,来说研究者可以基于注释好的参考转录组来进行RNA-seq分析。RNA-seq分析能单独作为转录组的profile,也可以和其他组学工具结合进一步拓展基因表达的分析。本文中,我们主要讲解RNA-seq的现有流程和工具,但不会详细地讲这些工具的使用,而是集中于对RNA-seq提供一个guideline。
Figure1:RNA-seq的概括
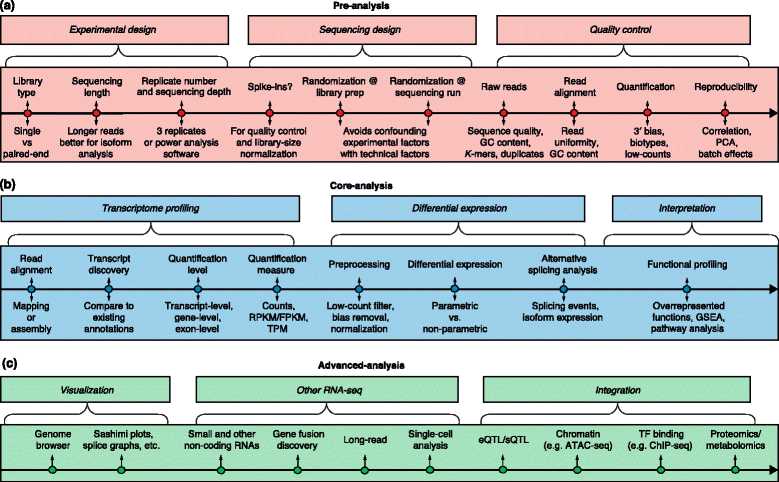
QC检测点:
1.下机数据的check:
为了检测测序错误,PCR错配,还有DNA或RNA污染,下机数据应该进行序列质量分析,GC含量分析,接头分析,k-mers过度分析,duplicated reads分析。可接受的duplication,k-mer或GC含量水平是经验性和种类特异性的,但同一批样品的这些参数应该保持一致。我们建议超过30%不一致的样品应该被丢弃。FastQC是一个用于分析IIIumina 数据的软件。一般说来,read 的质量会在3’末端开始下降,如果质量值太低,为了提高比对率,这些碱基应该被除掉。
2.reads alignment:
比对上的reads比例是一个重要的QC参数,这个参数能指示整个测序的准确性和是否有污染DNA的存在。比如一般认为常规的RNA-seq的reads会有70-90%比对到人类基因组上(具体取决于使用的mapper)。当使用转录组作为参考时,一般会将这个参数下调,因为参考转录组并没有覆盖所有的转录子,而multi-mapping reads将上调,因为不同的转录子可能分享同一个外显子。
另一个重要参数是reads map到外显子和参考组上的uniformity。如果在polyA富集的样品中,reads主要集中在转录子的3’末端,则这很有可能意味着在实验开始阶段得到的RNA质量是很low的。mapped reads的GC含量和PCR bises有关。有关的软件:Picard,RSeQC,Qualimap.
3.转录子定量:
当实际的转录子定量值确定后,为了验证归一化方法是否合适,应该用GC含量和基因长度偏差来检查定量值。如果参考转录组已经被很好的注释 ,那么研究者可以分析样品的生物成分,这可以表明RNA纯化步骤的质量。比如rRNA和sRNA应该不能被检测到在由polyA纯化制备的RNA文库中。常用软件:NOISeq,EDASeq。
Figure2:三种常规的RNA-seq分析策略
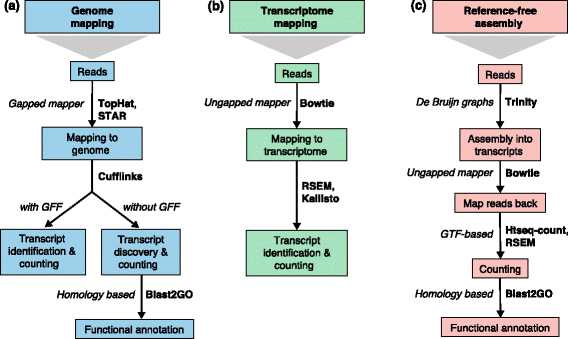
新转录子的发现:
在RNA-seq总,用短reads发现一个新的转录子是IIIumina技术的一个最大挑战。短reads很少能横跨多个剪切点,这使得直接发现full-length的转录子得十分困难。另外,很难确定新转录子的起点和终点。PE reads和更高的覆盖率能促进低表达量转录子的重构。Cufflinks , iReckon , SLIDE and StringTie 等现有方法利用已知的注释信息将mapped reads 归于可能的转录子。Gene发现工具如Augustus能将RNA-seq 的reads更好地整合到编码蛋白的转录子中,但对于非编码蛋白的转录子效果会更差。一般说来,从short reads里重构准确的转录子是很困难的,不同的方法得到的结果差异会很大。
转录子定量:
RNA-seq分析最常用的功能就是评估基因和转录子的表达。该功能主要是通过比对到转录组上的reads的数量来决定的,尽管有一些算法如Sailfish,不需要mapping,而是通过k-mer计算。定量的最简单方法是用软件统计mapped reads的raw counts,如HTSeq-count,featureCounts.gene-level定量主要是利用含有外显子的基因组坐标的GTF(gene transfer format)文件,并丢弃multireads。由于受到转录子长度,总reads数量和测序偏差等因素影响,raw counts是不足与比对不同样品的表达水平的。RPKM(reads per kilobase of exon model per million reads)是一种归一化的方法,它去除了转录子长度,库大小的影响。RPKM,FPKM(fragments per kilobase of exon model per million mapped reads),和TPM(transcripts per million)是最常用于RNA-seq基因表达值的参数。对于SE reads来说,RPKM和FPKM是等价的,FPKM能通过公式转化为TPM.
Cufflinks利用从maapper 如tophat得到的mapping to genome的数据来评估转录子的表达量,它用expectation-maximization的方法评估转录子的丰度。Cufflinks采纳了PE reads的优点,同时能用GTF的信息来确定转录子,或是能单独的从mapping data中进行转录子从头组装。
基因差异表达分析:
基因差异化表达要求基因表达能在不同样品中进行对比。RPKM,FPKM和TPM是利用归一化方法使得不同样品间能够进行对比。由于这些归一化方法是基于总的或有效的raw ounts,
所以当样品有heterogeneous transcript distributions时,它们的功能就有限了,因为高度表达,和差异表达会引起count分布的偏移。将这些因素考虑在内的归一化方法有TMM,DESeq,PossionSeq,UpperQuartile,它们忽略了高度表达和差异表达的特性。
标签:工具 统计 http fusion uart oba 没有 mil 流程
原文地址:http://www.cnblogs.com/timeisbiggestboss/p/7094855.html