标签:max cti with contain xpl eth 跳过 ogr cut
R需要包:mice包;AUC包或pROC包
需要知识:logistic回归;SVM;GAM;缺失值多重插补法;拟合效果ROC、AUC
In cancer studies, a question of critical interest is the progression of cancer. Here we consider a study on cancer progression.
The dataset contains the following variables:
Response: cancer development, which is a binary variable indicating whether there is any new cancer development;
Covariates: we have six covariates: size, stage, age, treatment type, dose I, dose II. Among them, stage and treatment type are categorical; the reminders are continuous.
Our question: in clinical practice, whether it is possible to predict cancer development using the six covariates we measure.
数据示例如下
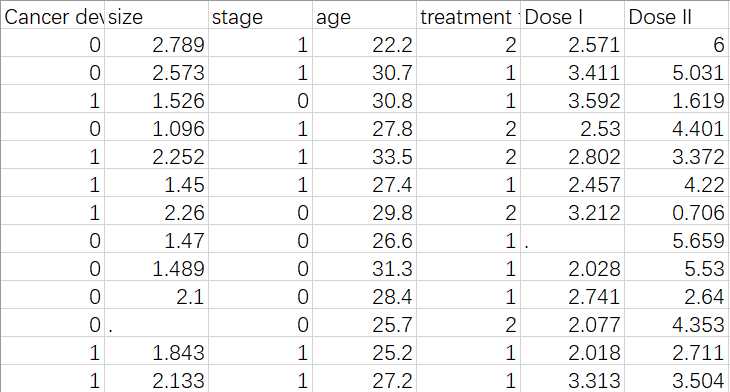
Cancer development: Yes (coded as 1) or No (coded as 0)
size: tumor size (which is measured with the diameter of tumor)
stage: tumor stage (categorial)
age: age (continuous)
treatment type: different type of treatment
Dose I: dose of drug A
Dose II: dose of drug B
".": missing measurements. You can assume MAR.
cancer <- read.csv("cancer-pred.csv", stringsAsFactors = FALSE)
cancer <- cancer[ , 1:7]
str(cancer)
查看数据结构,200个观测值和7个变量,并且存在缺失值。
由于数据集中缺失值由点号代替,故读取后变量为字符型,需将其转化为数值型。
无法通过设置read.csv中参数na.strings="."实现跳过转化步骤,因为最后两个变量含有小数点。
cancer <- sapply(cancer, as.numeric) #警告信息是由于点号转化为NA值
cancer <- as.data.frame(cancer) #转化为数据框更适用于建模
colnames(cancer) <- c("develop", "size", "stage", "age", "type", "Dose.I", "Dose.II")
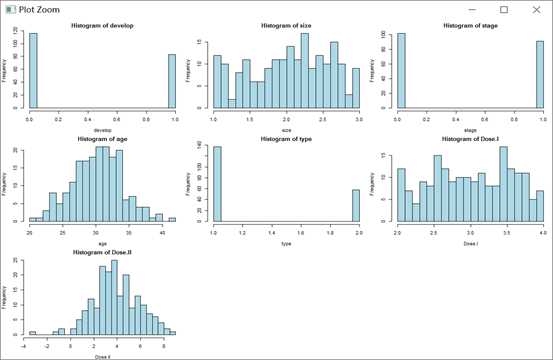
变量箱线图、柱状图、连续变量相关图,图片仅展示柱状图
old.par <- par(mfrow=c(3, 3))
apply(cancer, 2, boxplot)
par(old.par)
par(mfrow=c(3, 3), mar=c(4, 4, 2, 0.5))
for (j in 1:ncol(cancer)) {
hist(cancer[ , j], xlab=colnames(cancer)[j],
main=paste("Histogram of", colnames(cancer)[j]),
col="lightblue", breaks=20)
}
par(mfrow=c(1, 1))
pairs(~size+age+Dose.I+Dose.II, data=cancer)
对于缺失值的处理方法有几种,参考《R语言实战》
此处处理方法有三种:删除缺失值,平均值众数替代法,多重插补法。
# #缺失值删除法
# cancer <- na.omit(cancer)
# #平均数众数替代法
# cancer.mean <- sapply(cancer[c(2, 4, 6, 7)], mean, na.rm=TRUE)
# cancer.table <- sapply(cancer[ , c(1, 3, 5)], table)
# cancer[which(is.na(cancer[ , 2])), 2] <- round(cancer.mean[1], 3)
# cancer[which(is.na(cancer[ , 4])), 4] <- round(cancer.mean[2], 1)
# cancer[which(is.na(cancer[ , 6])), 6] <- round(cancer.mean[3], 3)
# cancer[which(is.na(cancer[ , 7])), 7] <- round(cancer.mean[4], 3)
# for(i in c(1, 3, 5)){
# cancer[which(is.na(cancer[ , i])), i] <-
# as.numeric(rownames(cancer.table)[which.max(cancer.table[1])])
# }
# #多重插补法
imp <- mice(cancer, seed=1)
fit <- with(imp,glm(develop~., family=binomial, data = cancer))
pooled <- pool(fit)
cancer <- complete(imp,action=3)
cancer[ , c(3, 5)] <- sapply(cancer[ , c(3, 5)], as.factor) #部分变量转化为因子
将数据分为训练集(150个样本)和测试集(50个样本),用训练集拟合logistic模型,用得到的模型对测试集的因变量进行预测
将预测值与真实值比较得到错判矩阵、tpr、fpr,针对不同的cutoffs得到roc曲线并计算AUC值。
将以上步骤循环3000次处理,对3000个AUC取平均并进行t检验。
cancer.auc <- c(length=3000) #生成空向量以填充
for (i in 1:3000){
set.seed(i)
sample.i <- sample(1:nrow(cancer), 3/4*nrow(cancer))
cancer1 <- cancer[sample.i, ]; cancer2 <- cancer[-sample.i, ]
glm.can <- glm(develop~., data=cancer1)
# glm.new <- step(glm.can, trace=0) #trace=0不返回结果
cancer.pred <- predict(glm.can, family=binomial,
newdata = cancer2, type = "response")
cancer.roc <- roc(cancer.pred, as.factor(cancer2$develop))
cancer.auc[i] <- auc(cancer.roc)
}
mean(cancer.auc) #3000次AUC均值
t.test(cancer.auc, alternative = "greater", mu=0.5)
hist(cancer.auc, freq = F, breaks = 40)
lines(density(cancer.auc, bw=0.05), col="red", lwd=2)
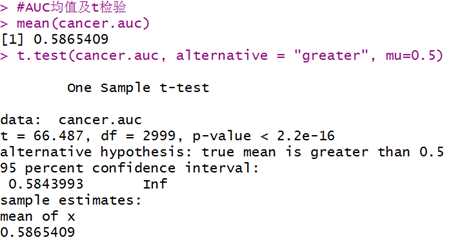
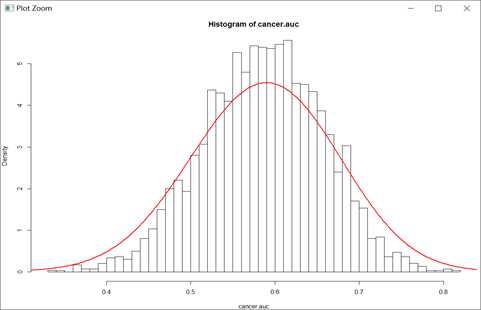
因变量为是否型,虽然t检验通过,但是预测效果并不好
下面使用支持向量机的方法
#SVM
cancer.auc <- c(length=3000)
for (i in 1:3000){
set.seed(i)
sample.i <- sample(1:nrow(cancer), 3/4*nrow(cancer))
cancer1 <- cancer[sample.i, ]; cancer2 <- cancer[-sample.i, ]
cancer.svm <- svm(develop~., data=cancer1kernel= "linear")
cancer.pred <- predict(cancer.svm, newdata = cancer2,
type="response")
cancer.roc <- roc(cancer.pred, as.factor(cancer2$develop))
cancer.auc[i] <- auc(cancer.roc)
}
mean(cancer.auc)
t.test(cancer.auc, alternative = "greater", mu=0.5)
尝试SVM四种核方法,即kernel参数“linear”、“polynomial”、“radial”、“sigmoid”
最后线性方法得到AUC均值最大,但也只有0.5837262,与logistic模型相比并不具有显著优势
#GAM
cancer.auc <- c(length=300)
for (i in 1:300){
set.seed(10*i)
# set.seed(3)
sample.i <- sample(1:nrow(cancer), 3/4*nrow(cancer))
cancer1 <- cancer[sample.i, ]; cancer2 <- cancer[-sample.i, ]
cancer.gam <- gam(develop ~ s(size, bs="cr") + stage +
s(age, bs="cr") + type + s(Dose.I, bs="cr") +
s(Dose.II, bs="cr"), family = binomial,
data=cancer1, trace=TRUE)
cancer.pred <- predict(cancer.gam, newdata = cancer2,
type="response")
cancer.roc <- roc(cancer.pred, as.factor(cancer2$develop))
# auc(cancer.roc)
cancer.auc[i] <- auc(cancer.roc)
}
mean(cancer.auc)
t.test(cancer.auc, alternative = "greater", mu=0.5)
GAM函数得到300次AUC均值为0.5694343,且GAM循环3000次较GLM运算速度相比很慢
Tips:
问题一:随机抽样,以下得到的试验集与测试集不是互补关系,会导致AUC上升。
set.seed(1) cancer1 <- cancer[sample(1:nrow(cancer), 3/4*nrow(cancer)), ] cancer2 <- cancer[-sample(1:nrow(cancer), 3/4*nrow(cancer)), ]
问题二:三种处理缺失值方法(缺失值剔除法、均值众数替代法、多重插补法)结果相差不大,差距在0.01左右。
问题三:DoseII数据出现小于0的值如果当作异常值处理删去结果为0.5770816,不处理结果为0.5873808,AUC降低了0.01,故放弃。
问题四:循环中使用step函数筛选不显著变量,AUC降低0.3左右,故放弃。
问题五:计算AUC值有两个包“AUC”和“pROC”,两个包roc函数参数输入方式不同。设立了随机数种子,但是二者得到结果测试集差0.01,试验集到小数点后20位才不一致,未确定是用AUC计算方式不同解释还是测试集出现问题解释。
若需原始数据试验可回复联系
标签:max cti with contain xpl eth 跳过 ogr cut
原文地址:http://www.cnblogs.com/lj0019/p/7197498.html