标签:represent 单线程 标准 全面 工作 中文 repr 建议 bsp
# 进入工作目录 cd example_PE250
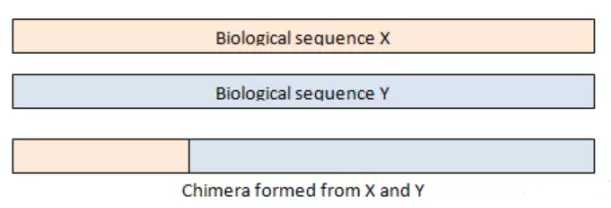
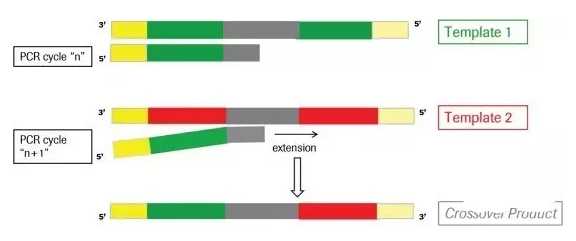
# 下载Usearch推荐的参考数据库RDP wget http://drive5.com/uchime/rdp_gold.fa # 基于RDP数据库比对去除已知序列的嵌合体 ./usearch10 -uchime2_ref temp/otus.fa -db rdp_gold.fa -chimeras temp/otus_chimeras.fa -notmatched temp/otus_rdp.fa -uchimeout temp/otus_rdp.uchime -strand plus -mode sensitive -threads 96
# 获得嵌合体的序列ID grep ‘>‘ temp/otus_chimeras.fa | sed ‘s/>//g‘ > temp/otus_chimeras.id # 剔除嵌合体的序列 filter_fasta.py -f temp/otus.fa -o temp/otus_non_chimera.fa -s temp/otus_chimeras.id -n # 检查是否为预期的序列数量2820 grep ‘>‘ -c temp/otus_non_chimera.fa
# 下载Greengene最新数据库,320MB wget -c ftp://greengenes.microbio.me/greengenes_release/gg_13_5/gg_13_8_otus.tar.gz # 解压数据包后大小3.4G tar xvzf gg_13_8_otus.tar.gz # 将OTU与97%相似聚类的代表性序列多序列比对,大约8min time align_seqs.py -i temp/otus_non_chimera.fa -t gg_13_8_otus/rep_set_aligned/97_otus.fasta -o temp/aligned/ # 无法比对细菌的数量 grep -c ‘>‘ temp/aligned/otus_non_chimera_failures.fasta # 1860 # 获得不像细菌的OTU ID grep ‘>‘ temp/aligned/otus_non_chimera_failures.fasta|cut -f 1 -d ‘ ‘|sed ‘s/>//g‘ > temp/aligned/otus_non_chimera_failures.id # 过滤非细菌序列 filter_fasta.py -f temp/otus_non_chimera.fa -o temp/otus_rdp_align.fa -s temp/aligned/otus_non_chimera_failures.id -n # 看我们现在还有多少OTU:975 grep ‘>‘ -c temp/otus_rdp_align.fa
# 重命名OTU,这就是最终版的代表性序列,即Reference(可选,个人习惯)
awk ‘BEGIN {n=1}; />/ {print ">OTU_" n; n++} !/>/ {print}‘ temp/otus_rdp_align.fa > result/rep_seqs.fa
# 生成OTU表
./usearch10 -usearch_global temp/seqs_usearch.fa -db result/rep_seqs.fa -otutabout temp/otu_table.txt -biomout temp/otu_table.biom -strand plus -id 0.97 -threads 10
# 结果信息 01:20 141Mb 100.0% Searching seqs_usearch.fa, 32.3% matched
# 默认10线程,用时1分20秒,有32.3%的序列匹配到OTU上;用30线程反而用时3分04秒,不是线程越多越快,分发任务也是很费时间的
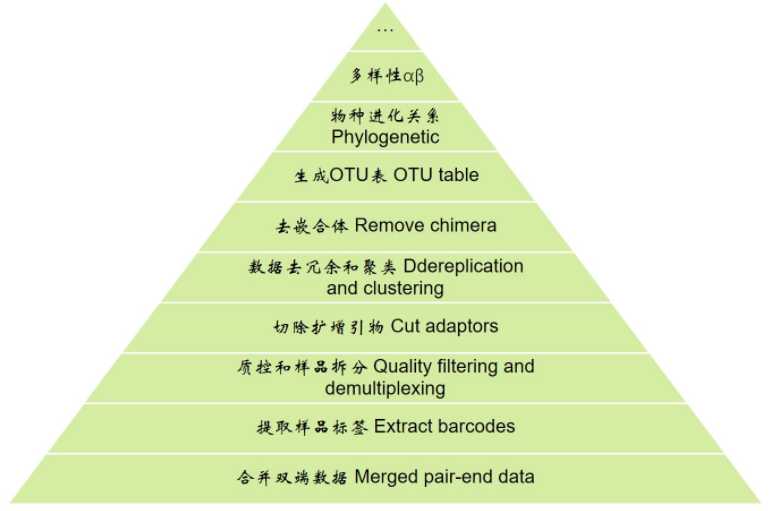
扩增子分析解读4去嵌合体 非细菌序列 生成代表性序列和OTU表
标签:represent 单线程 标准 全面 工作 中文 repr 建议 bsp
原文地址:http://www.cnblogs.com/freescience/p/7407455.html