标签:parameter mode basis nta journal ons lmos 图片 infer
An optimized molecular potential for carbon dioxide
Author: Zhigang Zhang, and Zhenhao Duan
Citation: The Journal of Chemical Physics 122, 214507 (2005); doi: 10.1063/1.1924700 View online: https://doi.org/10.1063/1.1924700
An optimized molecular potential model for carbon dioxide is presented in this paper. Utilizing the established techniques of molecular-dynamics and histogram reweighting grand canonical Monte Carlo simulations, this model is demonstrated to show excellent predictability for thermodynamic, transport, and liquid structural properties in a wide temperature-pressure range with remarkable accuracies. The average deviations of this new model from experimental data for the saturated liquid densities, vapor densities, vapor pressures, and heats of vaporization are around 0.1%, 2.3%, 0.7%, and 1.9%, respectively. The calculated critical point is almost pinpointed by the new model. The experimental radial distribution functions ranging from 240.0 to 473.0 K are well reproduced as compared to neutron-diffraction measurements. The predicted self-diffusion coefficients are in good agreement with the nuclear-magnetic-resonance measurements. The previously published potential models for CO2 are also systematically evaluated, and our proposed new model is found to be superior to the previous models in general.
V. CONCLUDING REMARKS
Optimization of the potential model has long been recognized as an important but nontrivial and even artistic job. For carbon dioxide, although a number of potential models have been proposed to reproduce different experimental properties, no one can be regarded as "perfect" to meet the insatiable appetites of different researchers. By noticing that none of the potential models available in the literature shows good predictability and transferability of volumetric properties and phase behaviors with sufficient accuracies simultaneously, it is our endeavor to find a better potential model for carbon dioxide in this study. On the basis of large number of computer simulations over the parameter spaces, the optimized potential model is expected to predict both volumetric properties and the phase coexistence envelope with improved accuracies.
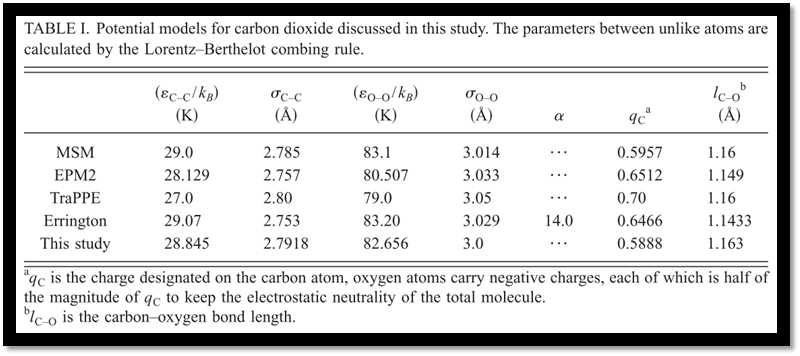
Along with the optimized potential in this study, four other popular site-site potential models for carbon dioxide (EPM2,5 MSM,8,11,20 TraPPE,12 and the model proposed by Errington6) have been comprehensively evaluated. For the volumetric properties of condensed phases with temperatures from 223.0 to 1073.15 K and pressures less than 100.0 MPa, our model generally gives the best predictions with relative errors lowered by about 1.0% as compared with the model of Errington and by 0.5% as compared with the other three models (Fig. 2). As pressures increased, the exponent-6 model of Errington shows better accuracies of about 0.5%. Our model gives a little larger deviation of about 1.0%. MSM enlarges the deviations to about 1.5% and EPM2 goes further to about 2.0%. TraPPE gives an even larger relative error of about 3.0%, which implies the failure of TraPPE model at high pressures (Figs. 3 and 4).
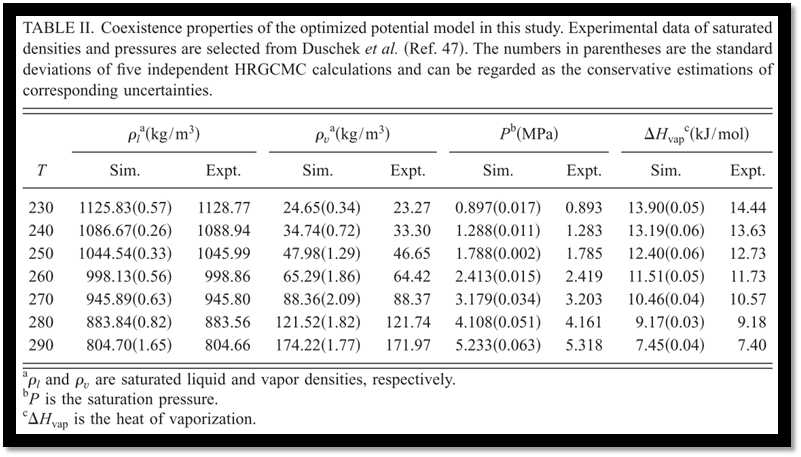
Among the five potential models examined in this study, our model gives the best predictions of phase behaviors. The saturated liquid densities, vapor densities, vapor pressures, and heats of vaporization are predicted with remarkable accuracies around 0.1%, 2.3%, 0.7%, and 1.9%, respectively. In particular, the critical point has been accurately reproduced. EPM2 shows comparable predictions but a systematical underestimation of about 0.7% for the saturated liquiddensities at lower temperatures has been observed, as compared with 0.1% of our model. MSM reproduce the experimental phase behaviors with slightly larger deviations. While the predictions of saturated liquid densities are very good, the model proposed by Errington only gives a less accurate description of saturated vapor densities, which reaches to about 10.3% at 290.0 K and this would result in a noticeable deviation of the critical point. According to our calculations, TraPPE only gives a rough description of the phase behaviors and significantly overestimates the critical point (Tables II–VI and Fig. 5).
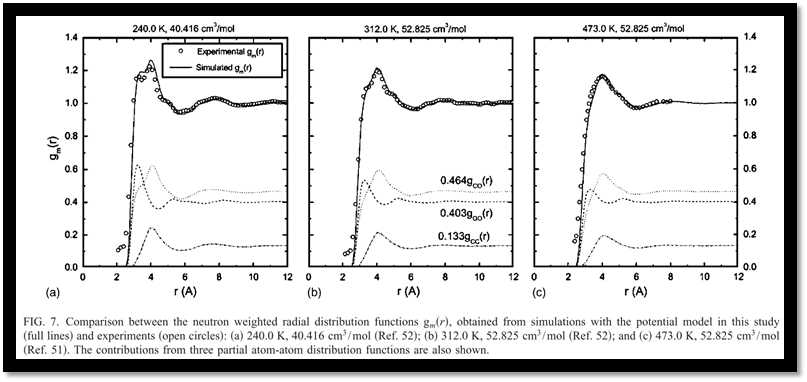
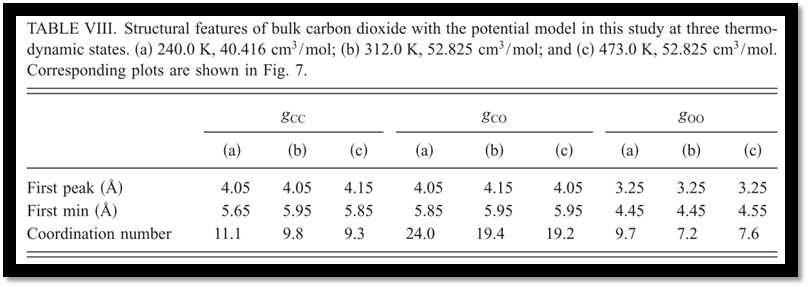
Subsequently, the structural properties have been studied with the optimized potential. Compared with neutron diffraction measurements, the overall agreements between the experiments and simulation results with our model are remarkable (Fig. 7). A further analysis of the radial distribution functions reveals the effects of the temperature and density on the bulk structures, and the boundary of the first coordination shell of carbon dioxide has been defined accordingly. With a cylindrical coordination system introduced (Fig. 8), the T-shape configurations are found to be predominant but are gradually distorted as temperature increases and density decreases for the sake of thermal fluctuations (Fig. 9).
Finally, we give a brief discussion of the simulated dynamic property of self-diffusion coefficient. Our model gives the best prediction from 273.0 to 373.0 K (Fig. 10), while EPM2 shows better accuracies of self-diffusion coefficients at lower temperatures. At higher temperatures, both models give comparable predictions. The other models are inferior in the self-diffusion coefficient predictions.
标签:parameter mode basis nta journal ons lmos 图片 infer
原文地址:https://www.cnblogs.com/Simulation-Campus/p/9038795.html