标签:ber 比例 功能 类的方法 聚类 inf 添加 个数 微软
一:导入R包及需要画热图的数据
library(pheatmap)
data<- read.table("F:/R练习/R测试数据/heatmapdata.txt",head = T,row.names=1,sep="\t")
二:画图
1)pheatmap(data)#默认参数
2)pheatmap(data,clustering_distance_rows = "correlation")#聚类线长度优化
3)pheatmap(data,scale="column")#按列均一化,"row","column" or "none"默认是"none"
4)pheatmap(data,scale="row")#按行均一化,"row","column" or "none"默认是"none"
5)pheatmap(data,display_numbers=T,number_format="%.2f",number_color="red",fontsize_number=8)#是否在每一格上显示数据,及其数据格式,大小及其颜色
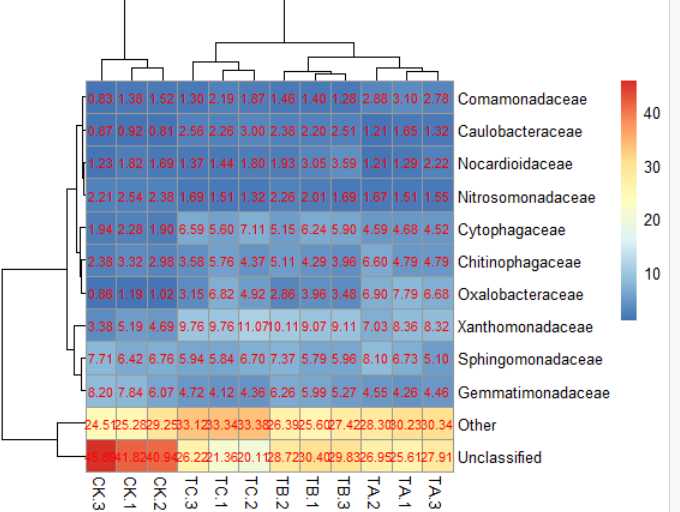
6)pheatmap(data,cellwidth = 50,cellheight= 14)#格子大小
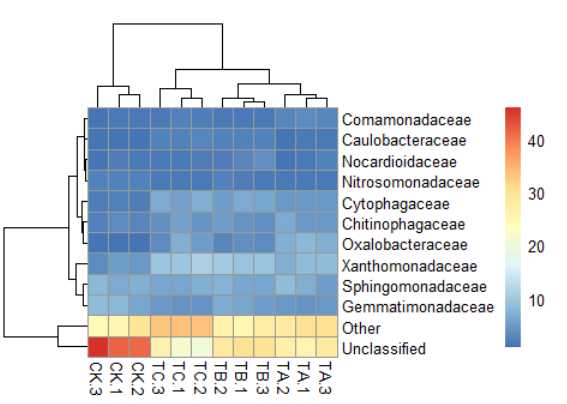
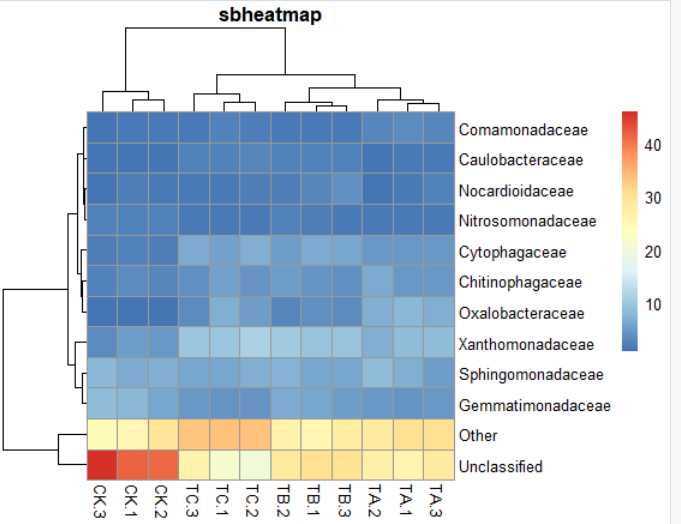
7) pheatmap(data,main="sbheatmap")#标题
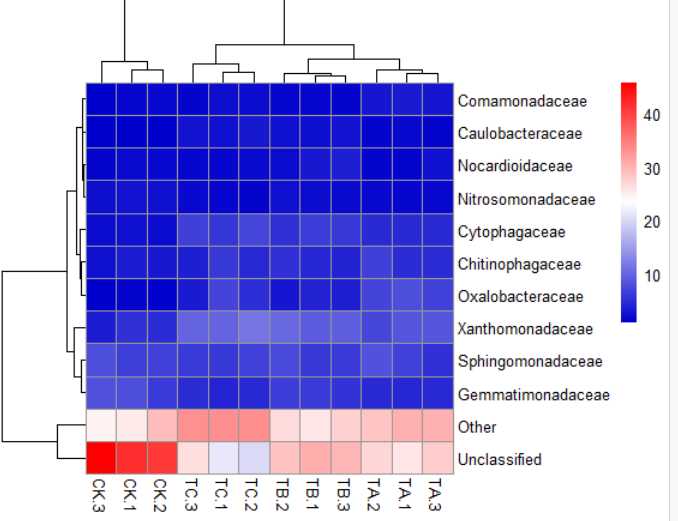
8)pheatmap(data,color = colorRampPalette(c("MediumBlue","white","red"))(256))#颜色
9)pheatmap(data,clustering_distance_rows = "correlation",scale="column",display_numbers=T,number_format="%.1f",number_color="black",
fontsize_number=8,cellwidth = 14,cellheight= 14,color = colorRampPalette(c("MediumBlue","white","red"))(256))

一、热图工具的参数与原理
(1)关于均一化的方法原理:
热图的核心思想是:使用渐变的颜色来代表数值的变化,以便其更加直观。使用RNA-seq的表达量数据绘制热图,最大的问题是不同基因的表达量差异过于巨大。例如:基因A的平均表达量是10,000(所有样本的表达量都在5,000~15,000),而基因B的平均表达量是100(所有样本的表达量都在50~500间波动)。那么问题来了,因为它们数值差异过大,根本不在一个数量级上,那么就很难在一张热图中使用合理的颜色标尺来反映两个基因在不同样本的表达量变化。
例如在下图中,横坐标代表样本,纵坐标代表基因。由于第一个基因的表达量非常高(相对其他基因),如果表达量不做任何处理,图中只显示这个基因表达量极高(红色),其他基因的表达量较低(黄色)。但到底第一个基因在不同样本间的表达量是如何变化了,就无法显示了。
图1 不合理的热图
所以在热图中,我们通常会对基因的表达量做一个归一化的处理。具体的做法,就是将每个基因的表达量减去这个基因在所有样本中表达量的均值,然后除以其标准差。这个处理也叫标准正态化,或Z-score处理(感兴趣的同学可以自己百度一下)。
这个处理非常巧妙,每一个基因在所有样本中的表达量被等比例缩放。这样处理后,每一个基因在所有样本的表达量,都变成了均值为0,标准差为1的一组值。以上的样本,被如此处理后,效果如下图。因为所有样本的表达量都在1个数量级的水平了,所以使用一套颜色配色体系,就可以很好地展示所有基因在不同样本的变化规律。你可以注意一下图右侧的图例标尺:数值的变动在-2~2之间。所以也有用户问过,怎么看起来不像表达量啊?的确不是表达量,是Z-core处理后的表达量,在0附近分布。
图2 合理归一化处理后的热图
(2)均一化的参数选择
如同刚刚我们所说的,热图的均一化目的是将贫富差距过大的基因,拉到同一个数量级。而通常在画热图的时候,1个基因在不同样本的表达量会在行的方向上分布。所以,我们的均一化处理将按行处理。所以这种情况下,我们的均一化参数选择:row。
当然,如果你的表达量表中,同一个基因的表达量在列的方向上分布,自然这个参数就要选择 column了。
注意要点(非常重要):
1)当数据中,某一行完全相同的时候,标准差=0,那么理论上是无法使用以上的Z-score公式进行均一化(公式中标准差是分母,不能为0)。所以,如果你的数据中包含某个基因(行方向)数值在所有样本完全相同的情况,OS-tools会自动将这一行删除。
2)如果你的数据只有两列,那么使用按行归一化的话,图形将很丑陋,如下图:
图3 略丑陋的双列热图
原因是当每行只有两个数值的时候,任何两个不同的数值标准正态均一化后,都会变成-1和1。所以,会产生上述的图形(只有两种颜色)。面对这种类型的数据,建议直接计算每行两个数值的倍数的log2值,然后使用OS-tools画单列的热图。
当然, 单列热图也可以使用R语言的pheatmap包绘制,并通过一个函数控制0点的位置,在另一个R语言绘图的主题帖中也有介绍。
(3)聚类的原理
聚类的方法很多。这里我就不花大量篇幅去解释了。我们用R语言pheatmap包,默认情况下将利用表达量信息计算两两样本间的欧氏距离,然后利用欧式距离实现样本的聚类。
如同上图的聚类效果,简单说来,在基因完成归一化处理后,如果我们对行聚类(在上图中也就是对基因聚类),那么基因的顺序就会被重排。表达规律比较相似的基因将会被排在一起。表达模式差异越大的基因,则会远离。类似的,如果对列聚类(在上图中也就是对样本聚类),那么样本的顺序将会被重排,表达模式比较相似的样本自动会被归为一类。
如上图,共有11个样本的约40个基因被用于绘制热图。从聚类的效果来看,11个样本可以归为两类,其实就是对应病人和正常人。40个基因也被归为两类,分别是病人组上调或下调表达的基因。
(4)聚类参数什么情况下使用
聚类的本质就是重排序,所以我们应该按照实际情况选择是否聚类。
a)需要聚类的情况:
需要对样本或基因按照表达模式分类,那么请选择聚类。例如,上图中需要对正常人和病人利用基因表达量进行分类,那么的确应该选择聚类。
另外,聚类的结果就是相似的东西被排布在一起,所以聚类后的图形也更加有序和美观。
b)不需要聚类的情况:
在某些情况下,我们只是需要使用热图来直观呈现基因在样本中的变化规律,而样本的顺序是我们提前定义好的,那么则要考虑将聚类功能关闭。
例如,在以下的热图中,选择了对行(基因)聚类,不对列(样本)聚类。那是因为作者希望通过聚类,将表达模式相似基因归为一类在图中展示,所以基因聚类选择yes。而样本(列)是作者提前排好序的,是小鼠三个组织在6个发育阶段的样本。因为样本是提前排好序的,当然作者不希望这个顺序被打乱,所以列选择不聚类。
备注:图中的分类标签,必须使用R包 pheatmap绘制热图才能添加。
图4 基因聚类但样本不聚类的例子。
还有两个方向都不聚类的例子。例如在下图中,X轴是1个实验处理后0h、5h、10h的样本,是作者提前排好序的。本意是想呈现相关基因在梯度时间水平的变化规律。当然,作者不想这个顺序被重新排布了,所以列方向的聚类选择:no。在y轴方向,这些基因也是作者提前按照其所属的基因家族排过序的,当然也不想其顺序被打乱,所以行聚类也是选择no。
图5 两个方向都不聚类的例子
(5)其他参数
工具中的其他参数还包括:
颜色选择:选择绘制热图的色系。考虑到绿红的色系,对红绿色盲来说区分有些困难,某些杂志不接受绿红色系。建议用户使用蓝红灯其他渐变色系;
字体大小:当热图样本、基因数太多的时候,可以通过减少字体大小来保证正常显示;
格子高、宽:主要为了美观而调整;
格子上是否显示数字:是否将表达量的数值写在格子中,就看用户自己选择了。
画出格子边界:如果相邻的格子颜色相似,可以通过画出边界来提高区分度。在格子数较少的时候,建议画出格子边界会更加美观。
用R包中heatmap画热图
标签:ber 比例 功能 类的方法 聚类 inf 添加 个数 微软
原文地址:https://www.cnblogs.com/djx571/p/9152627.html