标签:说明符 分区 注意 src pac tor 进一步 map padding

ref是参考序列,Read r001/1和 r001/2组成read pair,r003是嵌合体(chimeric read) ,r004表示 split alignment事件
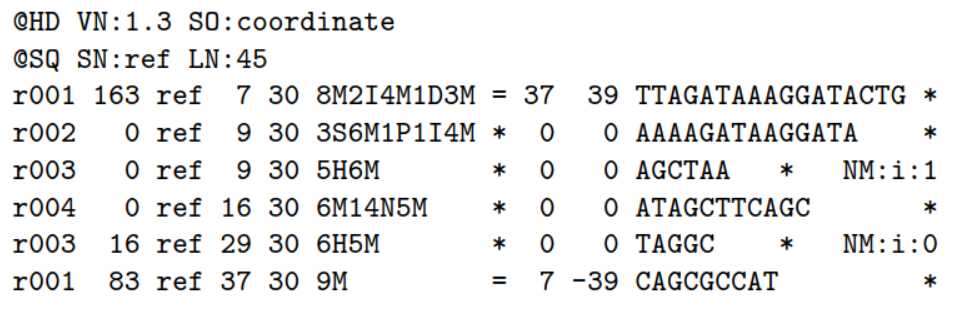
这11列内容的解释:
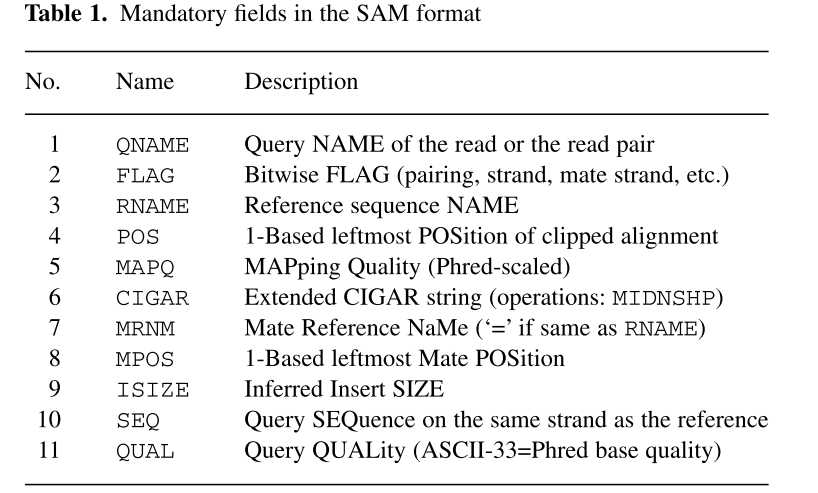
由此我们可以看到,SAM是由两部分组成:分为标头注释信息(header section)和比对结果(alignment section)。标头信息可有可无,都是以@开头,用不同的tag表示不同的信息,主要有:
@HD,说明符合标准的版本、对比序列的排列顺序(这里为coordinate)
@SQ,参考序列说明 (SN:ref,LN 是参考序列的长度)
@PG,使用的比对程序说明(这里没有给出)
比对结果部分(alignment section)每一行表示一个片段(segment)的比对信息,包括11个必须的字段(mandatory fields)和一个可选的字段,字段之间用tag分割。必须的字段有11个,顺序固定,根据字段定义,可以为’0‘或者’*‘,这11个字段是:
1)QNAME:比对片段的(template)的编号;
2)FLAG:位标识,template mapping情况的数字表示,每一个数字代表一种比对情况,这里的值是符合情况的数字相加总和;进一步学习可查看https://broadinstitute.github.io/picard/explain-flags.html
3)RNAME:参考序列的编号,如果注释中对SQ-SN进行了定义,这里必须和其保持一致,另外对于没有mapping上的序列;
4)POS:比对上的位置,注意是从1开始计数,没有比对上,此处为0;
5)MAPQ:mappint的质量;
6)CIGAR:简要比对信息表达式(Compact Idiosyncratic Gapped Alignment Report),其以参考序列为基础,使用数字加字母表示比对结果,比如3S6M1P1I4M,前三个碱基被剪切去除了,然后6个比对上了,然后打开了一个缺口,有一个碱基插入,最后是4个比对上了,是按照顺序的;
7)RNEXT:下一个片段比对上的参考序列的编号,没有另外的片段,这里是’*‘,同一个片段,用’=‘;
8)PNEXT:下一个片段比对上的位置,如果不可用,此处为0;
9)TLEN:Template的长度,最左边得为正,最右边的为负,中间的不用定义正负,不分区段(single-segment)的比对上,或者不可用时,此处为0;
10)SEQ:序列片段的序列信息,如果不存储此类信息,此处为’*‘,注意CIGAR中M/I/S/=/X对应数字的和要等于序列长度;
11) QUAL:序列的质量信息,格式同FASTQ一样
1 read是pair中的一条(read表示本条read,mate表示pair中的另一条read)
2 pair一正一负完美的比对上
4 这条read没有比对上
8 mate没有比对上
16 这条read反向比对
32 mate反向比对
64 这条read是read1
128 这条read是read2
256 第二次比对
512 比对质量不合格
1024 read是PCR或光学副本产生
2048 辅助比对结果
M: match/mismatch
I :插入 insertion(和参考基因组相比)
D: 删除 deletion(和参考基因组相比)
N: 跳跃 skipped(和参考基因组相比)
S: 软剪切 soft clipping ,(表示unaligned)
H: 硬剪切 hard clipping (被剪切的序列不存在于序列中)
P: 填充 padding(表示参考基因组没有,而reads里面含有位点)
bam文件是Sam 文件的二进制压缩格式,保留了与sam 完成相同的内容信息。SAM/BAM 文件可以是未排序的,但是按照坐标(coodinate)排序可以线性的监控数据处理过程。samtools可以用来转化bam/sam文件,可以merg,sort aligment,可以去除duplicate,可以call snp及indels.
标签:说明符 分区 注意 src pac tor 进一步 map padding
原文地址:https://www.cnblogs.com/djx571/p/9495388.html