标签:名称 输出 .com 自己 sem sof 使用 python环境 资源
1)背景
生物信息学研究经常涉及计算或提取基因的各种特征,如基因ID作图,GC含量计算和不同类型的基因长度,通过操纵基因模型,这些模型通常以GTF格式注释,可从ENSEMBL或GENCODE数据库获得。这种计算对于后续分析是必不可少。 然而,专门用于直接从GTF文件分析各种模式的基因模型的软件包尚未公开。虽然 BioMart在一定程度上可以来执行某些功能,但是它依赖于数据库查询并且有时会很慢。 此外,用户需要熟悉字段名称BioMart后端数据库中的表格可能也不方便。
因此,GTFtools(用Python实现,不依赖于任何非python第三方软件),一个独立的命令行软件,它提供了一组从基因模型中提取特征的功能。 它不依赖于任何现有的生物信息学工具,易于安装和使用。GTFtools为促进常规生物信息学分析提供了一种新的工具。
2)用法
因为是在python环境下执行,参数是通过‘argparse’ 包进行传递,因此需要安装该包。GTFtools 使用 GTF file (ENSEMBL or GENCODE) 作为输入文件, 输出文件格式用户可以指定bed或者bed-like格式。目前实施的主要功能包括合并外显子的计算(基因异构体的均值,中位数和最大长度),UTR,TSS,基因符号-ID映射。通过与下游软件bedtools连用,可以用来解决多种生物信息的提取。
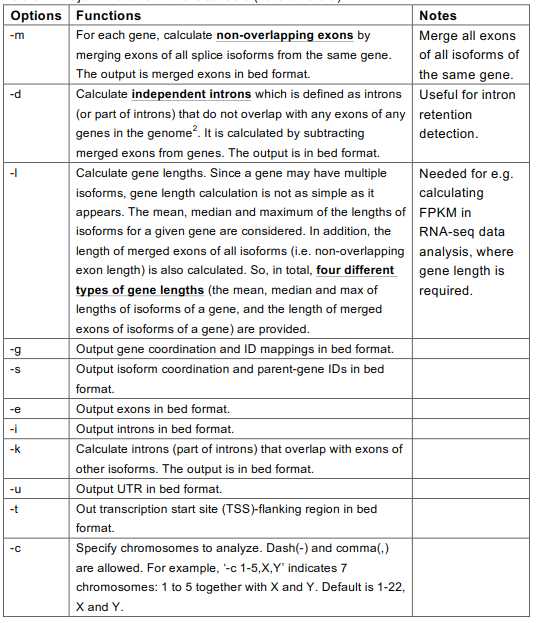
功能参数:
3)安装
1) wget http://www.genemine.org/codes/GTFtools_0.6.5.zip
2) unzip GTFtools_0.6.5.zip
3)echo ‘export PATH=/home/jxdong/biosoft/GTFtools_0.6.5:$PATH‘ >>~/.bashrc
4)alias gtftools=‘gtftools.py‘
5)source ~/.bashrc
4) 简单使用
软件里面自带测试数据demo.gtf
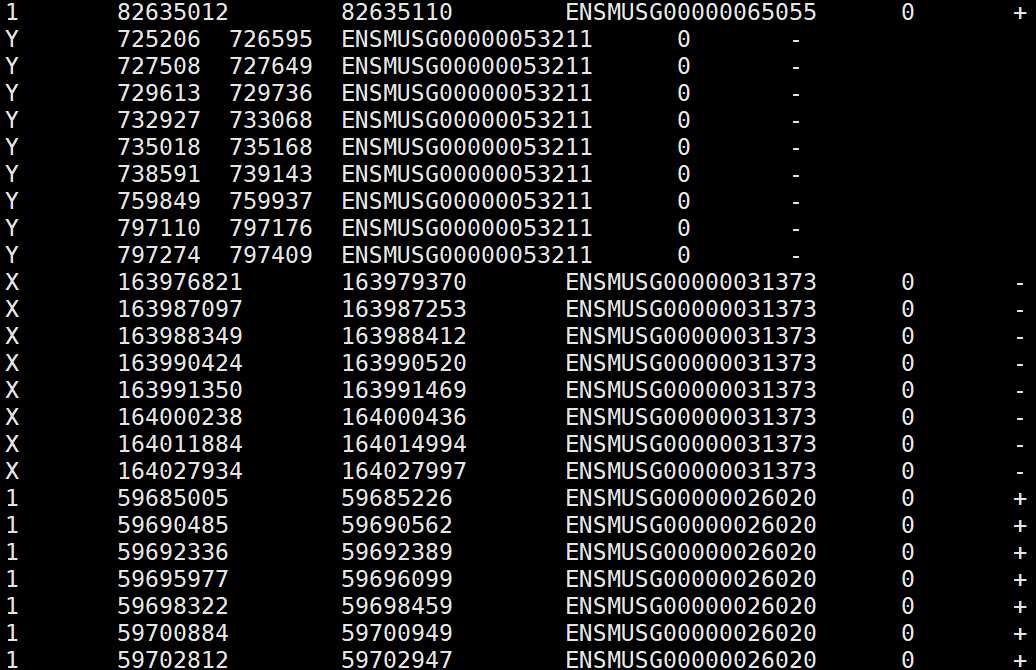
4.1)将extons进行merge
gtftools.py -m merged_exons.bed demo.gtf
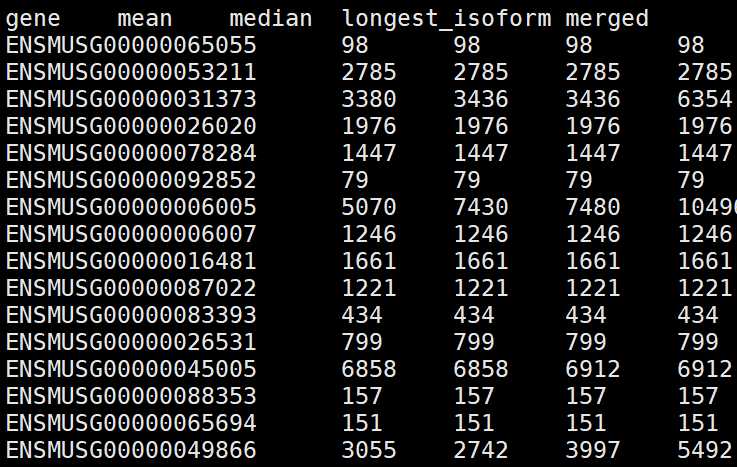
4.2)对基因进行统计
gtftools.py -l gene_length.bed demo.gtf
5)参考资源
GTFtools: a Python package for analyzing various modes of gene models
gtftools软件简单介绍(我自己不建议用,因为我发现不好用)
标签:名称 输出 .com 自己 sem sof 使用 python环境 资源
原文地址:https://www.cnblogs.com/djx571/p/9552541.html