标签:文件内容 file $0 9.png $$ 文件 $@ 参数 strong
来自知乎孟浩巍的“快速入门生物信息学的”Live,超棒的~
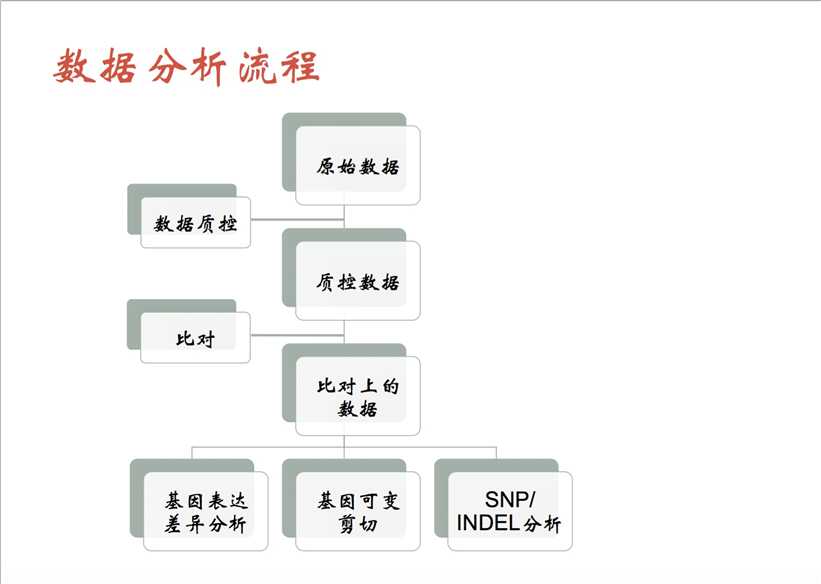
首先是质控部分,使用fastqc进行对结果分析。
对于Illumia二代测序的结果质控包括两个方面,去掉测序质量不好的序列,即Quality Control;二是需要去掉连在玻璃上的短的接头,cut adaptor。
![]()
-t 8表示调用8个核心去运算。
之后,对每一个序列文件都生成一个zip和一个html文件。
例如:
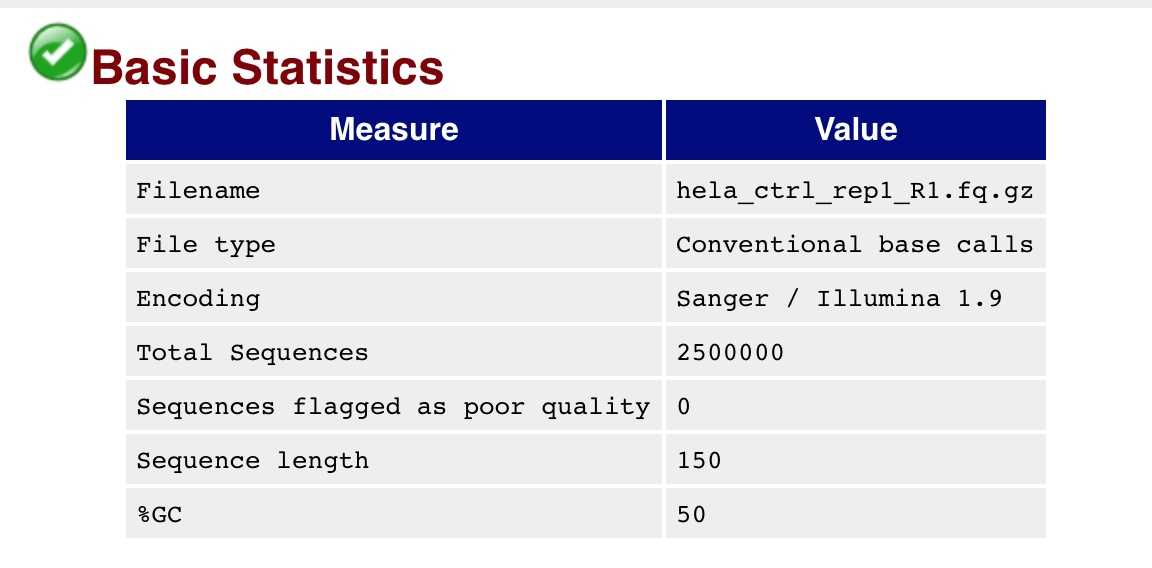
那么这2500000肯定是不同的基因,只不过这个机器的测序长度是150,所以所有的基因长度都是相同的。
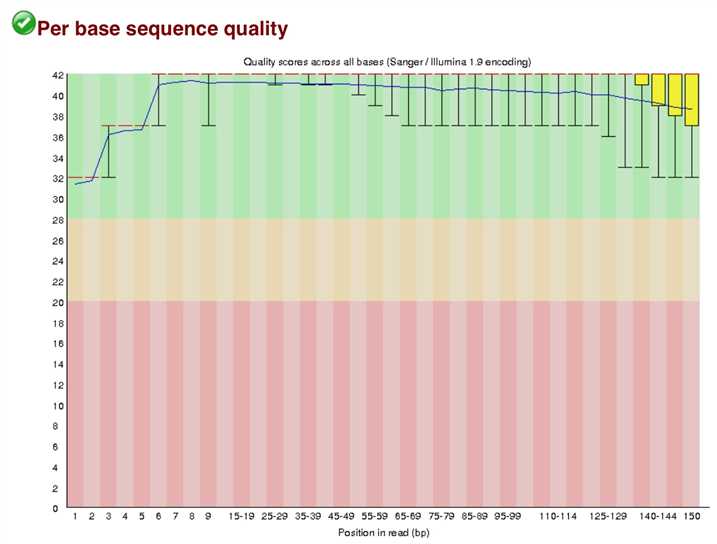
对250万reads,每个位点的Q做一个箱线图,要求箱线值最低点高于20%,否则需要将那部分切除。
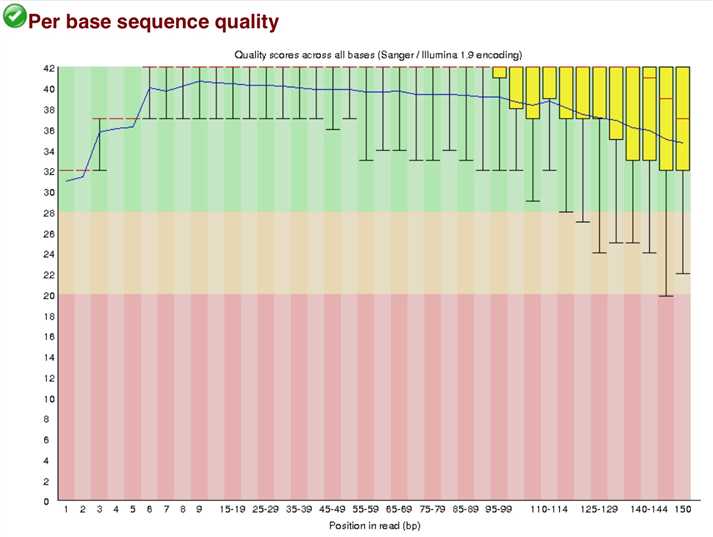
在145左右的的序列Q值较低测序不稳,所以这样的序列145之后的全不要了。
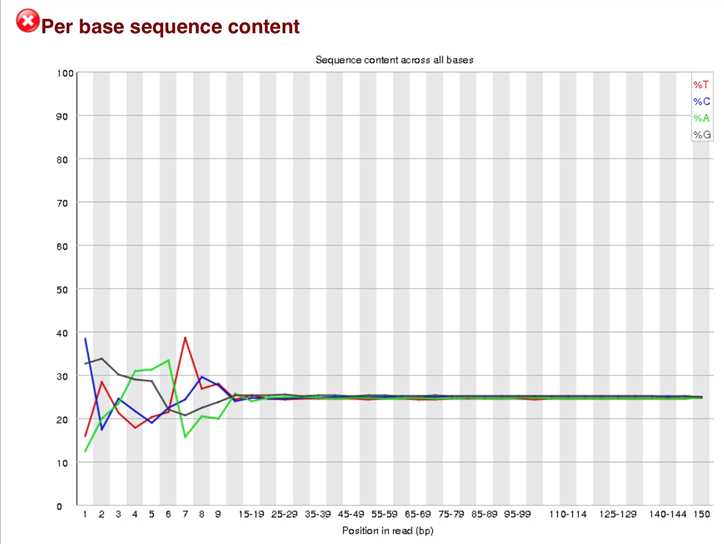
这个是GC含量图,通常A和T相同,C和G相同,但是前10bp不稳定,需要切除。
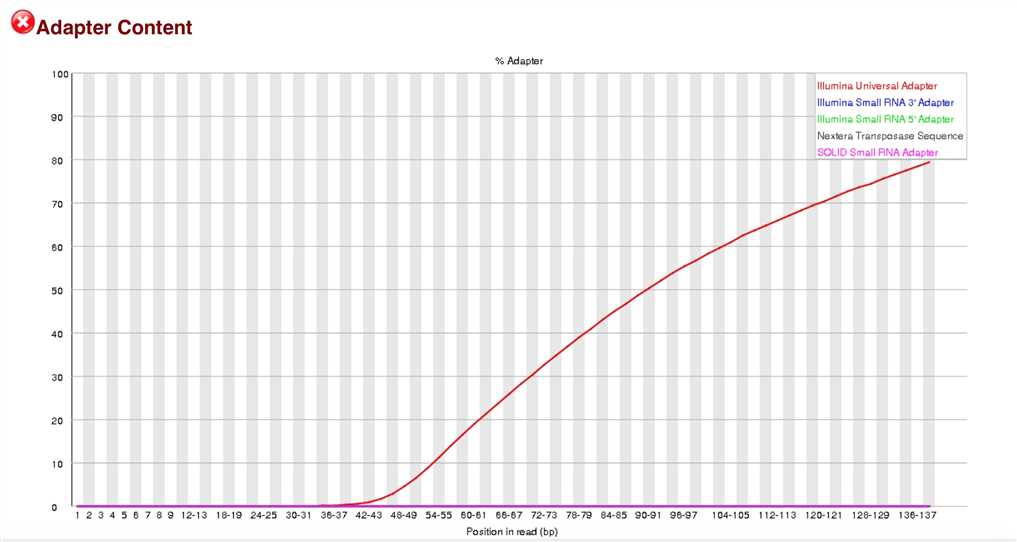
表示测序过程中测到的段序列的含量,横轴是1-150bp,纵轴是百分比。由于某种原因导致测的绝大多数都是测的adaptor。
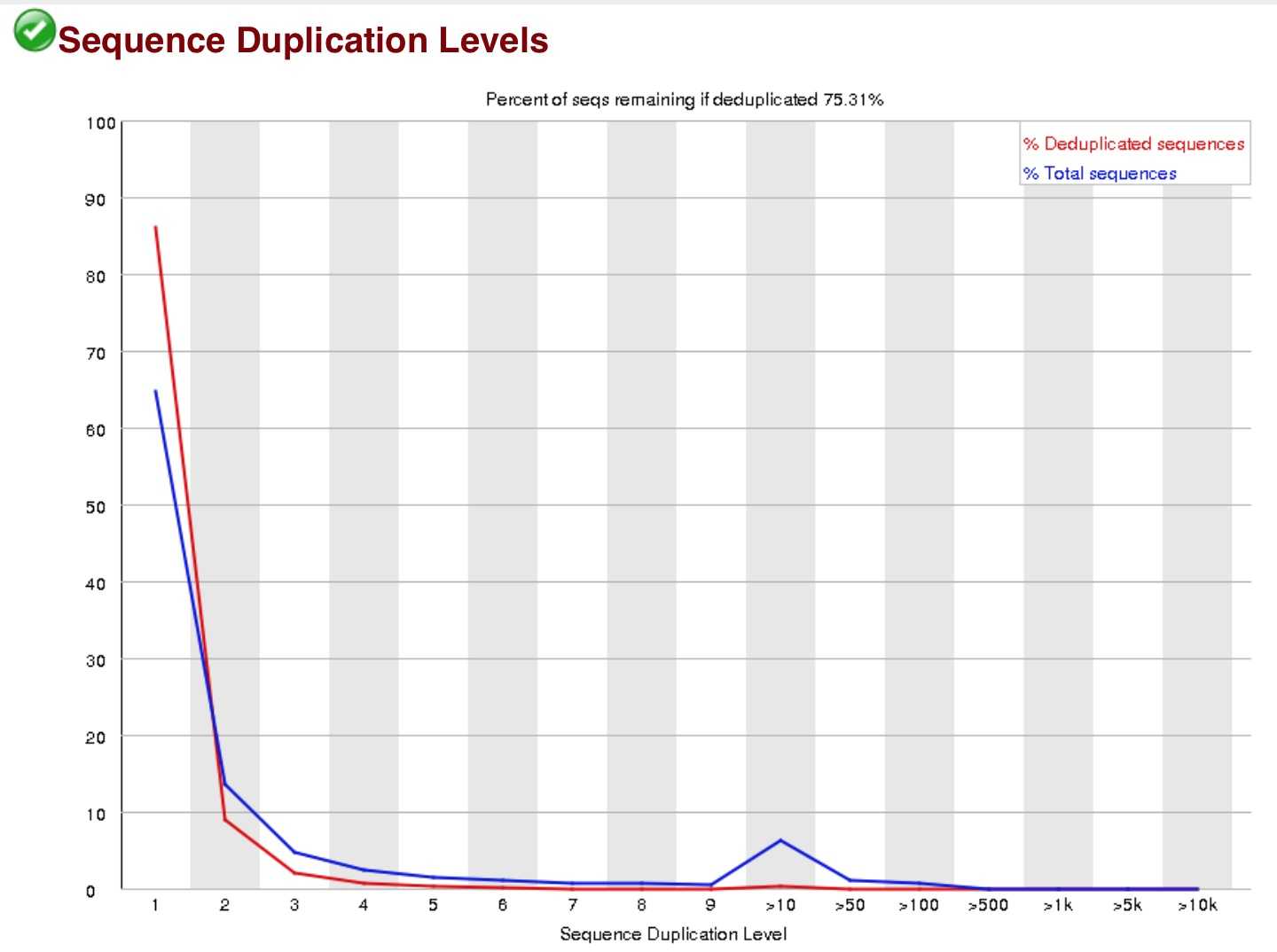
这个主要是衡量建库水平,建库中通常有6-8轮的PCR,但有时会出现过P的现象,当duplication过高的情况下,需要去dup。但是在RNA-seq里通常是不去dup的。
②接下来,使用fastx_trimmer去头去尾。
![]()
zcat $fastq_1 | fastx_trimmer -f 11 -l 140 -z -o $out_fastq_1 &
zcat解压缩,$fastq_1是输入的第一个文件,这个文件解压缩之后的结果给fastx_trimmer这个命令,
这个命令的参数-f是指first即保留的第一个bp(这里前10bp剪切掉了);last即保留的最后一个bp(保留到第140bp),-z是压缩命令,-o是输出到这个文件里。
//其中$:在bash里表示当前是普通用户;是变量引用操作符。a=10; echo $a会输出10。
③使用cutadaptor去掉两端的adaptor。
trimmer之后有一个去adaptor的过程,使用cutadaptor的软件,
![]()
nohup cutadapt --times 1 -e 0.1 -0 3
--quality-cutoff 6 -m 50 -a AGATCGGAAGAGC
-A AGATCGGAAGAGC -o $out_fastq_1
-p $out_fastq_2 $fastq_1 $fastq_2 > $log_file 2>$1 &
//其中nohup:不挂断地运行命令。
//2>$1:$1是传递给shell脚本的第一个参数;(转自:https://www.cnblogs.com/kaituorensheng/p/4002697.html)
$# 是传给脚本的参数个数
$0 是脚本本身的名字
$1 是传递给该shell脚本的第一个参数
$2 是传递给该shell脚本的第二个参数
$@ 是传给脚本的所有参数的列表
$* 是以一个单字符串显示所有向脚本传递的参数,与位置变量不同,参数可超过9个
$$ 是脚本运行的当前进程ID号
$? 是显示最后命令的退出状态,0表示没有错误,其他表示有错误
//times 1一条序列只去一次Adaptor;-e 0.1在匹配时可以有10%的错误率;-O 3 adaptor序列必须和测序序列有3个碱基以上的overlap才可以;常用6;-m 50如果处理之后低于50的话就扔掉序列,短序列测序质量可能不是很好;-a和-A是Illumina常用的通用引物,之所以输入两个,是因为我是一个双端测序的结果,需要对两个文件内容进行分别去除,-a对应Reads1,-A对应reads2,$fasrq_1和_2是上一步的输出;>最后是写入log文件
标签:文件内容 file $0 9.png $$ 文件 $@ 参数 strong
原文地址:https://www.cnblogs.com/BlueBlueSea/p/9820982.html
{kind=link}