标签:bsp sage 基本 重复 method 调整 mic cage 信号
RNA-seq表达谱与生物复制的差异表达分析。 实现一系列基于负二项分布的统计方法,包括经验贝叶斯估计,精确检验,广义线性模型和准似然检验。 与RNA-seq一样,它可用于产生计数的其他类型基因组数据的差异信号分析,包括ChIP-seq,Bisulfite-seq,SAGE和CAGE。
edgeR包是进行RNA-seq数据分析非常常用的一个R包。该包需要输入每个基因关于每个样本的reads数的数据,每行对应一个基因,每一列对应一个样本。edgeR作用的是真实的比对统计,因此不建议用预测的转录本。
归一化原因:
技术原因影响差异表达分析:
1)Sequencing depth:统计测序深度(即代表的是library size);
2)RNA composition:个别异常高表达基因导致其它基因采样不足
3)GC content: sample-speci?c e?ects for GC-content can be detected
4)sample-speci?c e?ects for gene length have been detected
注意:edgeR必须是原始表达量,而不能是rpkm等矫正过的。
>SFTSV_24vscontrol_circ<-read.table("edgeR_TPM_SFTSV_24vscontrol_circ.txt",header= T,stringsAsFactors = F)[,1:8] #读取基因表达量数据矩阵,包含6列样本(可分为C1和D1两组,每组各3个样本)共计1031行基因。
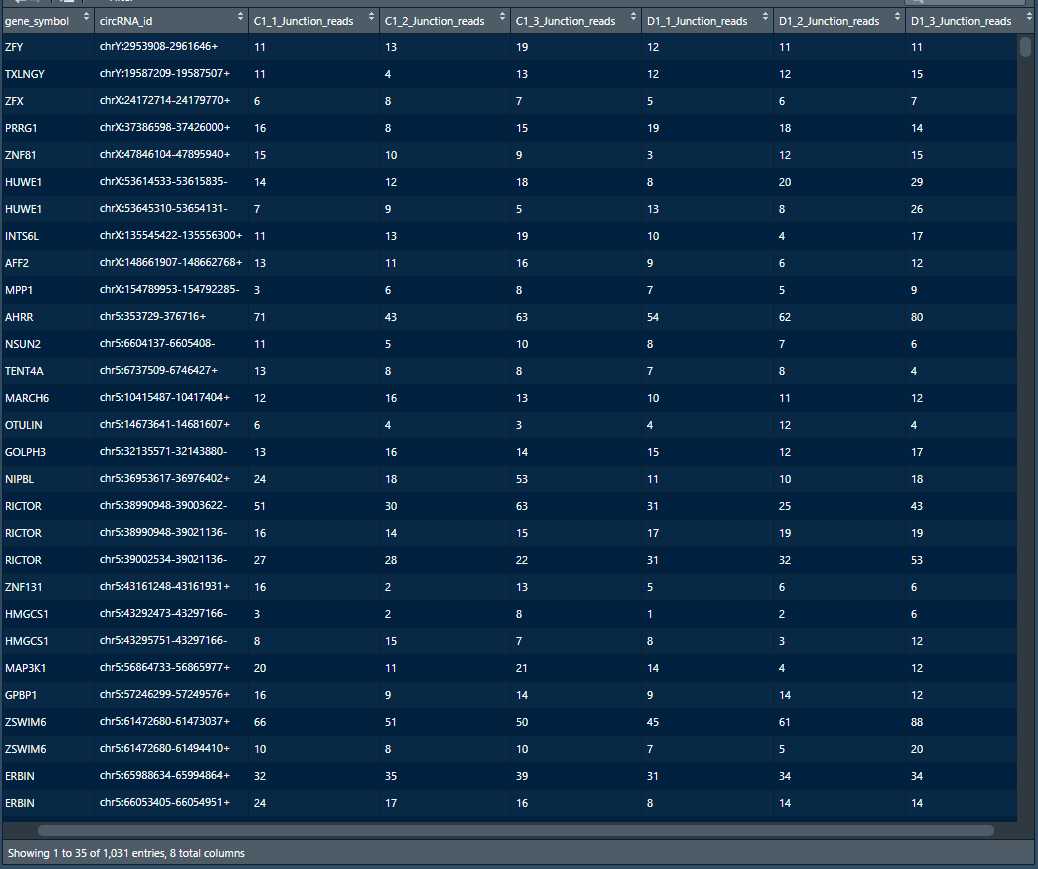
分为control组和SFTSV_24组,每组都为三个重复
>group<-c(rep("control",3),rep("SFTSV_24",3))
这里因为已经有rawdata的count文件,因此直接用DGEList()函数就行了,否则要用readDGE()函数
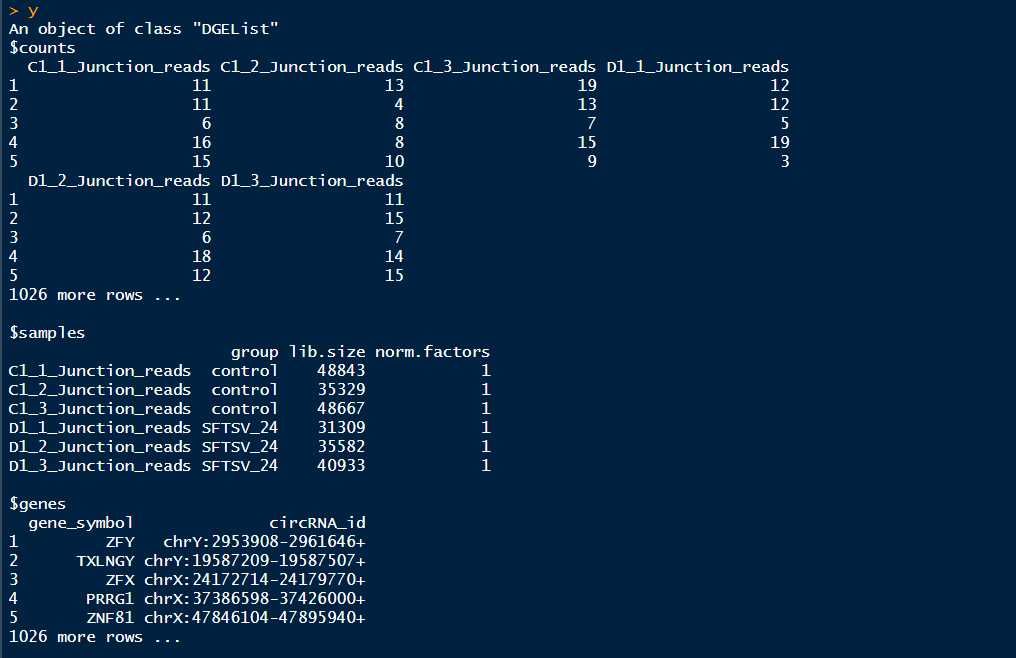
DGEList对象主要有三部分:
1、counts矩阵:包含的是整数counts;
2、samples数据框:包含的是文库(sample)信息。一共有4列,第一列为每个组的组名,第二列为group列,是上面构建的分组变量,第三列是lib.size列,如果不自定义lib.size,lib.size为每组记录的count之和,第四列为norm.factors列。
3、一个可选的数据框genes:gene的注释信息
###由于这里记录的是circRNA的back-spliced junction reads,这里的数据是取所有样本的circRNA的交集,所以这里的lib.size需要重新设定,设置为最开始的每个样本中所有circRNA的count之和###
>y<-DGEList(count=SFTSV_24vscontrol_circ[,3:8],group=group,genes=SFTSV_24vscontrol_circ[,1:2],lib.size = c(48843,35329,48667,31309,35582,40933)) #这里重新设置了lib.size
过滤:因为这里记录的是circRNA的count数,已经在分析的过程中过滤过,所以这里不进行数据的过滤。
如果是mRNA或者lncRNA的count数,需要对其的表达量进行过滤。对于在大多数样本中表达数量都很少的基因,需要进行过滤,这一步可以根据自己定义的标准过滤,edgeR推荐使用该包的CPM( count-per-million )值进行过滤,命令:
>keep <- rowSums(cpm(y)>1) >= 2#至少在两个样本里cpm大于1 >y <- y[keep, , keep.lib.sizes=FALSE]
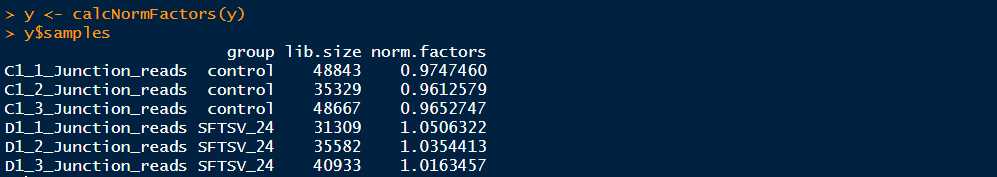
标准化:edgeR的标准化思想主要针对的是不同样本在建库时效应。这一点与RPKM不同,因为edgeR认为不同的基因对于所有样本的影响是相同的,所以不必考虑。因此为了消除这种建库时的效应,edgeR会更推荐你使用他的calcNormFactors函数,算出来的值叫做trimmed mean of M-values (TMM) ,命令为:
>y <- calcNormFactors(y) >y$samples
>plotMDS(y) #这里主要是通过图形的方式来展示实验组与对照组样本是否分得开,以及同一组内样本是否能聚的比较近,即重复样本是否具有一致性
> y <- estimateCommonDisp(y, verbose=TRUE) > y <-estimateTagwiseDisp(y) > plotBCV(y)

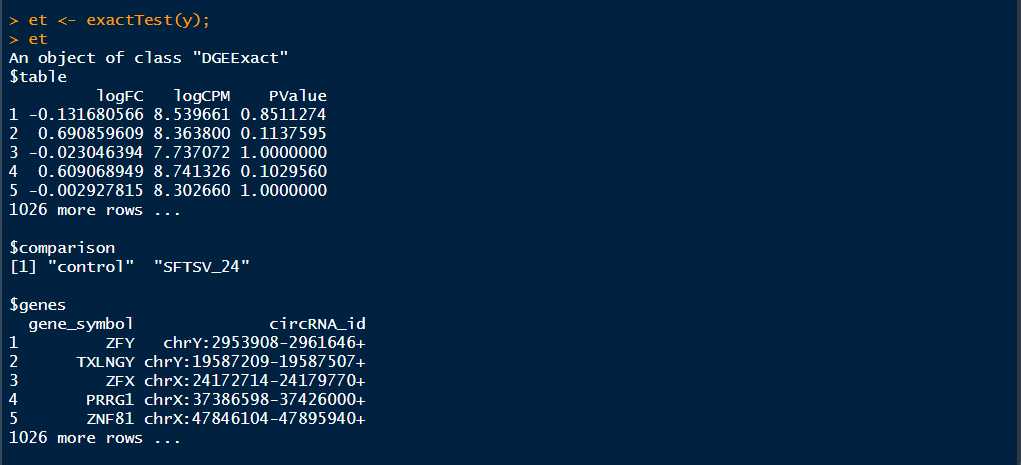
>et <- exactTest(y)
>top <- topTags(et) #这里可以设置topTags函数的参数,输入?topTags可以查看topTags的详细用法,可以设置参数n来调整显示的条目,默认是n=10,可以通过设置sort.by="logFC"来按照|logFC|的降序排序,设置p.value=0.05来筛选p-adjusted value<0.05的基因
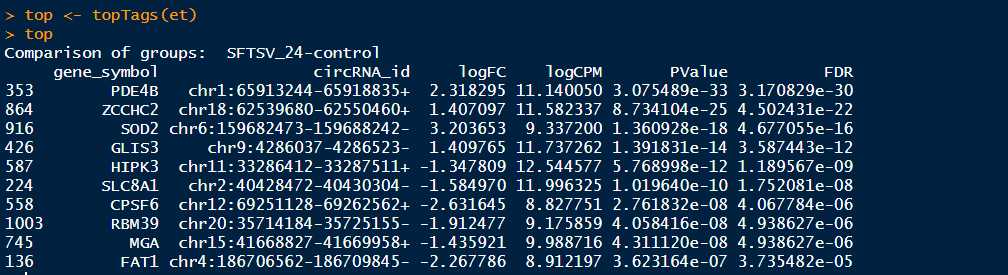
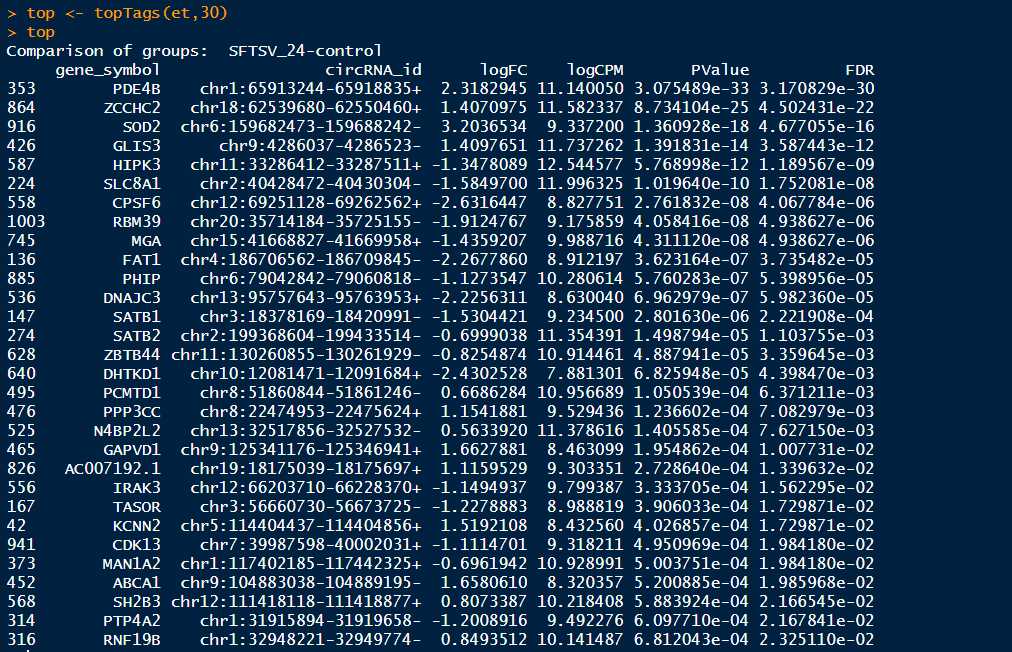
>summary(de <- decideTestsDGE(et,adjust.method = "BH", p.value = 0.05,lfc=1)) #默认的参数是adjust.method="BH",也可以设置为adjust.method="fdr",BH和fdr是等价的,p.value=0.05也是默认的参数,指的是筛选FDR小于0.05的基因,这里也可以设置lfc参数,默认的lfc为lfc=0,也可以根据自己需要设置,更多的函数参数可以输入?decideTestsDGE查看
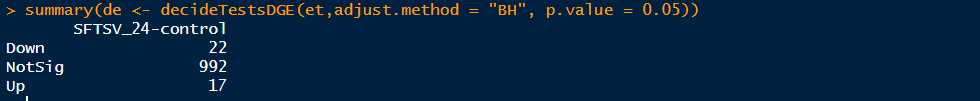

标签:bsp sage 基本 重复 method 调整 mic cage 信号
原文地址:https://www.cnblogs.com/yanjiamin/p/12059480.html