标签:数据量 延伸 single pad 原因 images 项目经验 就是 mesh
单细胞转录组
从研究方向看上,发育生物学、免疫、神经生物学、肿瘤是排名靠前的方向,这和我们平时遇到的高频研究方向基本吻合。另外,作为一个新兴的领域,10X 单细胞转录组检测到细胞多,数据庞大,信息复杂,对数据分析带来诸多困难,因此算法类的文章(Computational method)也高达76篇。
从物种上看,小鼠和人牢牢占据主流。毕竟人类医学研究还是生物领域的最大热门,小鼠也是头号模式动物。其他“飞禽走兽”已经慢慢都有涉及,但比较少的是植物(这里只有两例拟南芥的文章)。植物因为细胞壁的存在,制备单细胞悬液的难度更大,从而限制了大规模应用。不过这些困难也已经慢慢在摸索中被克服。
从组织类型上看,研究内容几乎涵盖了动物体内大部分组织器官,尤其在脑、血液、实体瘤、肺等四类样本发文的数量都已经超过50篇。所以,后续在人、小鼠领域没有任何实验设计,仅仅对此类已被大量研究的热门组织直接进行测序是发不了好文章的。所以,对已被大量文献报道的热门组织开展研究,个性化的实验设计尤为重要。当然,对于冷门的组织或者没有文献报道过的物种(例如大部分植物),只要成功测到数据,任何结果都是创新,则可以较少考虑复杂的实验设计问题。
在已发表的文献上看,截至2020年,10X单细胞转录组的文章依然很大比例发表在高分的主流期刊上。但这样的新技术红利不会一直持续下去,所以对于关注新技术的老师,还是早关注,早启动,早发文章才能保证有好的产出。

图2 10x单细胞转录组文章涉及的领域方向
(注意,分类上会有重复,比如研究方向涉及两个,所以细分之和会超过总数)
10x 免疫组库(VDJ-seq)
截至2020年5月,一共发文56篇。这是仅次于10X RNA-seq的热点方向,因为很多关心免疫细胞的老师会进行10X RNA-seq的时候,配对进行scVDJ-seq。但目前10X scVDJ-seq标准化试剂盒只针对人和小鼠,其他物种的用户如果想做只能自己去设计定制探针系统(显然难度比较大),这限制了其他动物利用该技术开展研究。10X scVDJ-seq因为通常需要先分类淋巴细胞(T/B细胞)然后进行检测,目前最多是对血液开展研究,其次是研究肿瘤浸润的淋巴细胞,其他组织则目前研究报道还比较少,不少空白还留着大家去补充。
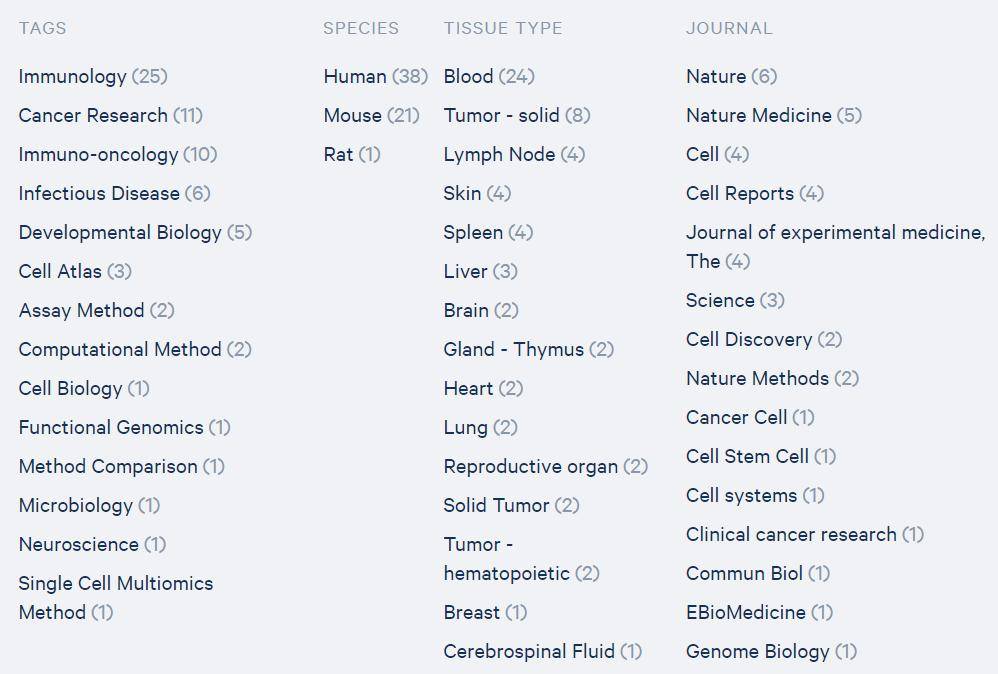
图3 10x单细胞免疫组文章涉及的领域方向
4. 空间转录组(ST-seq)
截至2020年5月,一共发文19篇。从发表文章上看,居然排名第一的是Scientific Report,实在太“辣眼睛”了:这么好的技术,暴殄天物啊。不过不用激动,这个技术其实直到2019年才被10X genomics公司收购,当年年底优化升级后推出。再此之前,这个技术所属的瑞典公司Spatial Tranomics一直不温不火的,发文章大部分也是一些瑞典的研究机构自己在玩。

图4 10x单细胞ATAC-seq文章涉及的领域方向
5. 10X ATAC-seq
截至2020年5月,一共发文12篇文章,数量还不多。而且,其中有近一半(5篇)是涉及生物信息分析方法探索的文章。这是由于对单细胞ATAC-seq这种信息庞大,噪音复杂的数据,应该如何分析还有很多值得探索的地方。
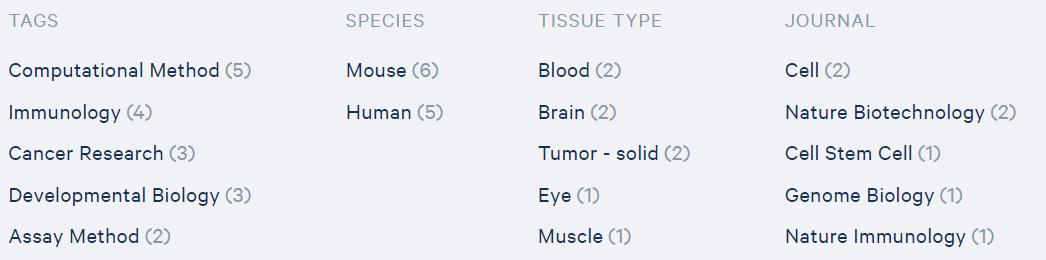
图5 10x单细胞ATAC-seq文章涉及的领域方向
从以上介绍,你可能已经发现,10X单细胞相关的转录调控组学技术目前主要围绕模式生物开展。那么10x单细胞技术是否可以研究非模式物种呢?
10X 单细胞技术可以检测哪些RNA以及应用于哪些物种
1. 10X单细胞技术是否需要参考基因组
以比较代表性的10X RNA-seq、VDJ-seq、ATAC-seq和ST-seq(空间转录组)来说。VDJ-seq受限于试剂只针对人和小鼠开发,因此其他物种目前无法开展商业化的服务。ATAC-seq作为检测基因组开放性的技术,其检测的区域大部分为非编码区,因此参考基因组不但必须要有,而且参考基因组的质量对ATAC-seq的影响非常大。
而对于RNA-seq或者ST-seq,本质上就是转录组,研究的目标分子是带ployA尾巴的RNA。因此,并非必须要有参考基因,只要有质量足够好的参考转录本就可以了。下来,我们重点剖析下10X RNA-seq和ST-seq的应用需求。
2. 10X RNA-seq/ST-seq可以检测哪些类型的RNA
从上文介绍,我们可以知道10X RNA-seq和ST-seq(空间转录组)依赖于围绕ployA结构开展扩增。那么我们分析一下10X RNA-seq/ST-seq可以检测哪些RNA。
(1)mRNA
由于真核生物mRNA都有ployA结构,所以理论上mRNA就是10X RNA-seq/ST-seq主要的检测目标。当然,由于只是扩增mRNA 3‘端或者5‘端的一小段用于定量,所以并不能能用于分析可变剪切。
(2)lncRNA
高等生物的LncRNA只有一部分有ployA结构(另外一部分自然没有),因此10X RNA-seq/ST-seq只能检测这些有ployA结构的lncRNA。另外,由于lncRNA表达量普遍比较低,而10X RNA-seq/ST-seq这类大规模单细胞/准单细胞测序的技术,对低丰度lncRNA分子的检测能力比较弱,因此结果中lncRNA的数量将比较少。
(3)其他RNA
近年来研究大热的环状RNA由于没有ployA结构,因此不在10X RNA-seq/ST-seq的检测范围内。同样的,其他类型的小RNA,例如miRNA,也是10X RNA-seq/ST-seq无法检测的。
3. 10X RNA-seq/ST-seq可以用于哪些物种研究
10X RNA-seq/ST-seq质上就是转录组测序。某个物种是否可以用10X RNA-seq/ST-seq开展转录组研究,需要考虑两个方面的问题:
(1)实验层面的问题
对于10X RNA-seq来说,主要考虑该物种是否可以制备单细胞或单细胞核悬液?大部分高等动物/植物的样本理论上都满足这个要求。而对于10X ST-seq主要要考虑该物种是否可以制作切片,以及切片中的组织是否可以被顺利解离释放RNA。对某些植物来说,在无法制作单细胞悬液的情况下,制作切片进行空间转录组测序或许是更可行的研究切入方式。这些技术的具体的实验方法,我们在后续章节讨论。
另外,细菌的细胞太小,且没有ployA结构,自然不适合10X genomics的检测。
(2)分析层面的问题
同常规RNA-seq一样,10X RNA-seq/ST-seq需要将测序数据比对到作为参考的基因组,才能实现对基因的定量。那么参考基因组是影响分析结果的主要问题。10X RNA-seq/ST-seq由于只对转录本的3‘端或者5‘端进行测序,然后通过比对参考基因组实现对RNA的定量。那么,这要求用于作为参考的基因组要有较高的质量。因为如果参考基因组组装质量差,基因注释不完整,那么会影响测序结果的比对以及基因定量。
基于参考基因组,我们可以分为3种情况:
1)参考基因组质量很高
比如,人类、小鼠、拟南芥、水稻等,参考基因组质量高,基因组注释都优化了很多版本了,开展10X RNA-seq/ST-seq分析自然没有问题了。
2)参考基因组质量值得怀疑
这10年来,基于二代测序组装技术的发展,很多非模式生物的参考基因组已经被发表。但实际上由于预算或急着发表等诸多因素,这些已经发表的基因组质量参差不齐。比如,很多基因组在注释的时候,只有CDS区注释,而缺乏5‘UTR或者3‘UTR区。而10X RNA-seq/ST-seq检测的是RNA的5’端或者3‘端序列,其实大部分就是5’UTR或者3‘UTR序列。如果参考基因组没有将UTR区域注释出来,自然就会影响测序结果的比对和定量。
所以,对哪些组装组质量较差的物种,如果比对率异常(比对在基因区的数据偏少),可以考虑人为对基因组注释文件的5’UTR区或者3‘UTR区进行延伸,这样可能会改善比对和定量的结果。另外,如果预算许可,可以考虑在实验设计中加入一些常规转录组或者3代全长转录组,用于优化参考基因组的注释(不过,10X RNA-seq/ST-seq这么贵的技术都用上了,好像也不会在乎多测几个常规转录组了吧)。
3)没有参考基因组
没有参考基因组当然没法做比对和定量,也就无法开展10X RNA-seq/ST-seq分析。对于没有参考基因组的物种,从而组装一个基因组费用比较高且周期比较长。对于无参考基因组的物种,如果老师很想进行10X RNA-seq/ST-seq研究,那么也可以考虑对转录组数据进行拼接,构建一个转录本参考用于10X RNA-seq/ST-seq数据的比对和定量。
但如果采用转录组de novo拼接构建转录组,一定要注意3个问题:
a)一定要使用三代测序进行转录组拼接而非二代测序
基于常规的二代测序结果的 de novo 拼接获得的转录本大部分是不完整的,大概率缺失UTR区的序列,所以基于常规二代测序拼接的 de novo 转录组参考序列集并不适合用于作为10X RNA-seq/ST-seq的参考库。唯一合适的方法应该是基于三代全长转录组测序技术进行 de novo 拼接,去获得完整的转录本全长序列,才适合作为10X RNA-seq/ST-seq的参考库。
b)三代转录组较低的基因检出率需要数据量做保障
我们做过的大量有参考基因组物种三代转录组测序数据表明,三代全长转录组对基因的检出率平均在40%(即基因组如果有2万个基因,但三代全长转录组平均只能检出8000个基因)。这主要原因三代全长转录组只有获得mRNA全长,被算一个有效检出的完整转录本。但在全部数据里,全长转录本所占的比例并不高,尤其对低丰度基因的转录本漏检较多。
为了保证三代全长转录组能够较多检测低丰度的转录本,以保证 de novo 拼接的转录组参考集涵盖更多的基因,可以考虑适当加大测序的数据量(现在三代测序也比较便宜了)。
c) de novo 参考转录组冗余度的影响
de novo 从头拼接的结果有一个比较麻烦的问题是序列冗余度比较大,即同一个基因的多个可变剪切同时被检测和拼接出来。这会导致10X genomics数据进行比对时,多重比对(即一条测序的reads会比对上多个转录本)比例比较大。而多重比对的reads在10X RNA-seq/ST-seq定量的时候,默认要被丢弃。
所以,对于 de novo 拼接来源的转录本需要适当进行去冗余处理,从而减少多重比对的影响,提高数据量的有效率。在无参考转录组 de novo 拼接方面,基迪奥有非常丰富的项目经验。在已有的案例中,我们已经证明了无参考转录组 de novo 拼接结果在进行适当优化后,可以作为10X RNA-seq/ST-seq的参考。
参考文献
[1] Svensson V, Vento-Tormo R, Teichmann S A. Exponential scaling of single-cell RNA-seq in the past decade[J]. Nature protocols, 2018, 13(4): 599.
[2] Rosenberg A B, Roco C M, Muscat R A, et al. Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding[J]. Science, 2018, 360(6385): 176-182.
[3] Macosko EZ, Basu A, Satija R, Nemesh J et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 2015 May 21;161(5):1202-1214
[4] CytoSeq: Fan H. C., Fu G. K. and Fodor S. P. (2015) Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science 347: 1258367
[5] Birey F, Andersen J, Makinson C D, et al. Assembly of functionally integrated human forebrain spheroids[J]. Nature, 2017, 545(7652): 54-59.
[6]单细胞在线课堂:https://www.omicshare.com/class/home/index/series?id=44
lncRNA
https://my.oschina.net/u/4503882/blog/4423031
https://www.sohu.com/a/397834526_278730
标签:数据量 延伸 single pad 原因 images 项目经验 就是 mesh
原文地址:https://www.cnblogs.com/emanlee/p/14955462.html