标签:des style blog http io color ar os 使用
http://blog.pluskid.org/?p=17
好久没有写 blog 了,一来是 blog 下线一段时间,而租 DreamHost 的事情又一直没弄好;二来是没有太多时间,天天都跑去实验室。现在主要折腾 Machine Learning 相关的东西,因为很多东西都不懂,所以平时也找一些资料来看。按照我以前的更新速度的话,这么长时间不写 blog 肯定是要被闷坏的,所以我也觉得还是不定期地整理一下自己了解到的东西,放在 blog 上,一来梳理总是有助于加深理解的,二来也算共享一下知识了。那么,还是从 clustering 说起吧。
Clustering?中文翻译作"聚类",简单地说就是把相似的东西分到一组,同 Classification(分类)不同,对于一个classifier ,通常需要你告诉它"这个东西被分为某某类"这样一些例子,理想情况下,一个 classifier 会从它得到的训练集中进行"学习",从而具备对未知数据进行分类的能力,这种提供训练数据的过程通常叫做?supervised learning?(监督学习),而在聚类的时候,我们并不关心某一类是什么,我们需要实现的目标只是把相似的东西聚到一起,因此,一个聚类算法通常只需要知道如何计算相似度就可以开始工作了,因此 clustering 通常并不需要使用训练数据进行学习,这在 Machine Learning 中被称作?unsupervised learning?(无监督学习)。
举一个简单的例子:现在有一群小学生,你要把他们分成几组,让组内的成员之间尽量相似一些,而组之间则差别大一些。最后分出怎样的结果,就取决于你对于"相似"的定义了,比如,你决定男生和男生是相似的,女生和女生也是相似的,而男生和女生之间则差别很大,这样,你实际上是用一个可能取两个值"男"和"女"的离散变量来代表了原来的一个小学生,我们通常把这样的变量叫做"特征"。实际上,在这种情况下,所有的小学生都被映射到了两个点的其中一个上,已经很自然地形成了两个组,不需要专门再做聚类了。另一种可能是使用"身高"这个特征。我在读小学候,每周五在操场开会训话的时候会按照大家住的地方的地域和距离远近来列队,这样结束之后就可以结队回家了。除了让事物映射到一个单独的特征之外,一种常见的做法是同时提取 N 种特征,将它们放在一起组成一个 N 维向量,从而得到一个从原始数据集合到 N 维向量空间的映射——你总是需要显式地或者隐式地完成这样一个过程,因为许多机器学习的算法都需要工作在一个向量空间中。
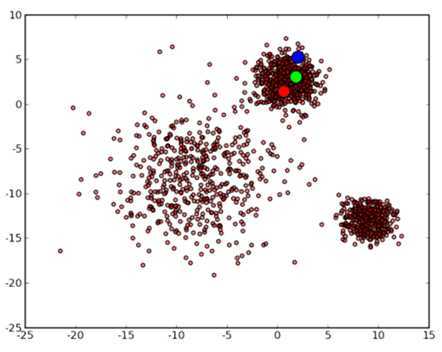
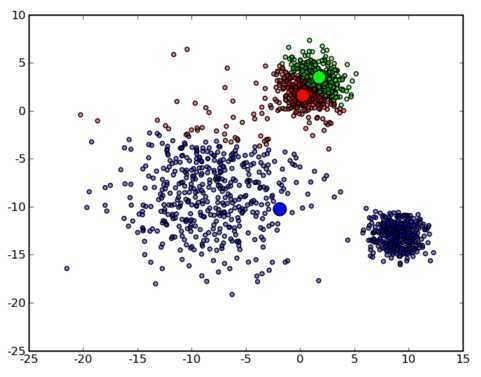
那么让我们再回到 clustering 的问题上,暂且抛开原始数据是什么形式,假设我们已经将其映射到了一个欧几里德空间上,为了方便展示,就使用二维空间吧,如下图所示:
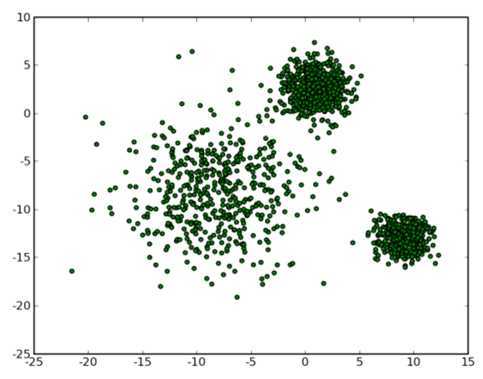
从数据点的大致形状可以看出它们大致聚为三个 cluster ,其中两个紧凑一些,剩下那个松散一些。我们的目的是为这些数据分组,以便能区分出属于不同的簇的数据,如果按照分组给它们标上不同的颜色,就是这个样子:
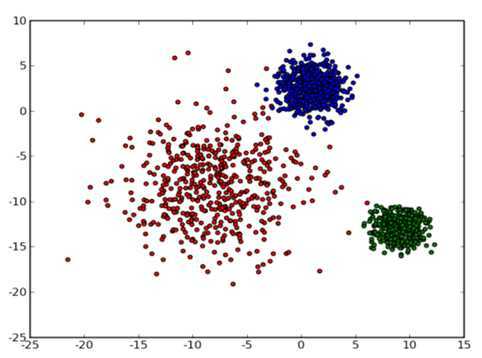
那么计算机要如何来完成这个任务呢?当然,计算机还没有高级到能够"通过形状大致看出来",不过,对于这样的 N 维欧氏空间中的点进行聚类,有一个非常简单的经典算法,也就是本文标题中提到的k-means 。在介绍 k-means 的具体步骤之前,让我们先来看看它对于需要进行聚类的数据的一个基本假设吧:对于每一个cluster ,我们可以选出一个中心点 (center) ,使得该 cluster 中的所有的点到该中心点的距离小于到其他 cluster 的中心的距离。虽然实际情况中得到的数据并不能保证总是满足这样的约束,但这通常已经是我们所能达到的最好的结果,而那些误差通常是固有存在的或者问题本身的不可分性造成的。例如下图所示的两个高斯分布,从两个分布中随机地抽取一些数据点出来,混杂到一起,现在要让你将这些混杂在一起的数据点按照它们被生成的那个分布分开来:
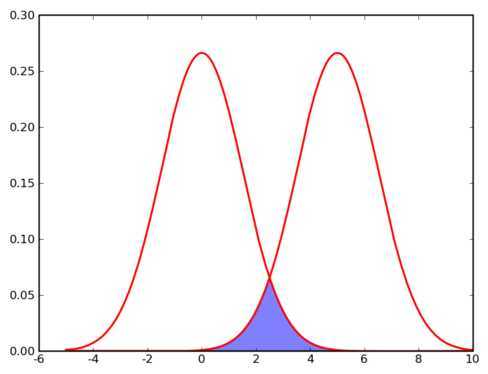
由于这两个分布本身有很大一部分重叠在一起了,例如,对于数据点2.5来说,它由两个分布产生的概率都是相等的,你所做的只能是一个猜测;稍微好一点的情况是2,通常我们会将它归类为左边的那个分布,因为概率大一些,然而此时它由右边的分布生成的概率仍然是比较大的,我们仍然有不小的几率会猜错。而整个阴影部分是我们所能达到的最小的猜错的概率,这来自于问题本身的不可分性,无法避免。因此,我们将 k-means 所依赖的这个假设看作是合理的。
基于这样一个假设,我们再来导出 k-means 所要优化的目标函数:设我们一共有 N 个数据点需要分为 K 个 cluster,k-means要做的就是最小化
这个函数,其中?在数据点n被归类到cluster k的时候为1,否则为0。直接寻找??和??来最小化??并不容易,不过我们可以采取迭代的办法:先固定??,选择最优的??,很容易看出,只要将数据点归类到离他最近的那个中心就能保证??最小。下一步则固定?,再求最优的?。将??对??求导并令导数等于零,很容易得到??最小的时候??应该满足:
亦即?的值应当是所有cluster k中的数据点的平均值。由于每一次迭代都是取到??的最小值,因此??只会不断地减小(或者不变),而不会增加,这保证了k-means 最终会到达一个极小值。虽然k-means并不能保证总是能得到全局最优解,但是对于这样的问题,像k-means 这种复杂度的算法,这样的结果已经是很不错的了。
下面我们来总结一下k-means 算法的具体步骤:
按照这个步骤写一个 k-means 实现其实相当容易了,在?SciPy?或者 Matlab 中都已经包含了内置的 k-means 实现,不过为了看看 k-means 每次迭代的具体效果,我们不妨自己来实现一下,代码如下(需要安装?SciPy?和?matplotlib) :
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 | #!/usr/bin/python ? ? from __future__ import with_statement import cPickle as pickle from matplotlib import pyplot from numpy import zeros, array, tile from scipy.linalg import norm import numpy.matlib as ml import random ? ? def kmeans(X, k, observer=None, threshold=1e-15, maxiter=300): N = len(X) labels = zeros(N, dtype=int) centers = array(random.sample(X, k)) iter = 0 ? ? def calc_J(): sum = 0 for i in xrange(N): sum += norm(X[i]-centers[labels[i]]) return sum ? ? def distmat(X, Y): n = len(X) m = len(Y) xx = ml.sum(X*X, axis=1) yy = ml.sum(Y*Y, axis=1) xy = ml.dot(X, Y.T) ? ? return tile(xx, (m, 1)).T+tile(yy, (n, 1)) - 2*xy ? ? Jprev = calc_J() while True: # notify the observer if observer is not None: observer(iter, labels, centers) ? ? # calculate distance from x to each center # distance_matrix is only available in scipy newer than 0.7 # dist = distance_matrix(X, centers) dist = distmat(X, centers) # assign x to nearst center labels = dist.argmin(axis=1) # re-calculate each center for j in range(k): idx_j = (labels == j).nonzero() centers[j] = X[idx_j].mean(axis=0) ? ? J = calc_J() iter += 1 ? ? if Jprev-J < threshold: break Jprev = J if iter >= maxiter: break ? ? # final notification if observer is not None: observer(iter, labels, centers) ? ? if __name__ == ‘__main__‘: # load previously generated points with open(‘cluster.pkl‘) as inf: samples = pickle.load(inf) N = 0 for smp in samples: N += len(smp[0]) X = zeros((N, 2)) idxfrm = 0 for i in range(len(samples)): idxto = idxfrm + len(samples[i][0]) X[idxfrm:idxto, 0] = samples[i][0] X[idxfrm:idxto, 1] = samples[i][1] idxfrm = idxto ? ? def observer(iter, labels, centers): print "iter %d." % iter colors = array([[1, 0, 0], [0, 1, 0], [0, 0, 1]]) pyplot.plot(hold=False) # clear previous plot pyplot.hold(True) ? ? # draw points data_colors=[colors[lbl] for lbl in labels] pyplot.scatter(X[:, 0], X[:, 1], c=data_colors, alpha=0.5) # draw centers pyplot.scatter(centers[:, 0], centers[:, 1], s=200, c=colors) ? ? pyplot.savefig(‘kmeans/iter_%02d.png‘ % iter, format=‘png‘) ? ? kmeans(X, 3, observer=observer) |
代码有些长,不过因为用 Python 来做这个事情确实不如 Matlab 方便,实际的 k-means 的代码只是 41 到 47 行。首先 3 个中心点被随机初始化,所有的数据点都还没有进行聚类,默认全部都标记为红色,如下图所示:
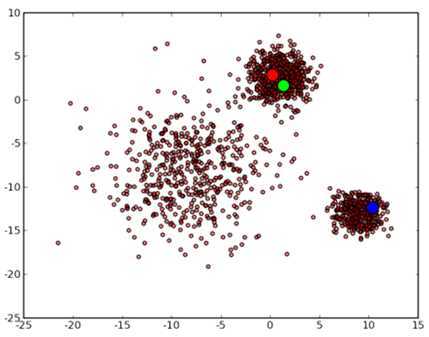
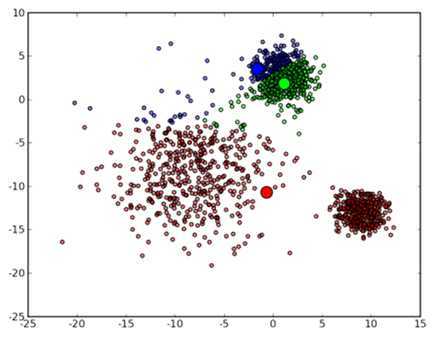
然后进入第一次迭代:按照初始的中心点位置为每个数据点着上颜色,这是代码中第 41 到 43 行所做的工作,然后 45 到 47 行重新计算 3 个中心点,结果如下图所示:
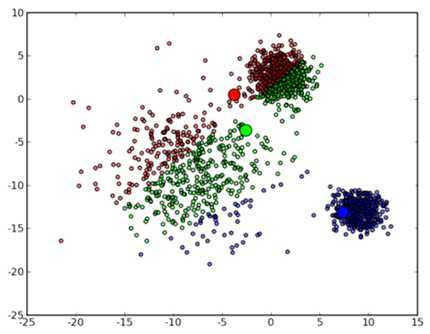
可以看到,由于初始的中心点是随机选的,这样得出来的结果并不是很好,接下来是下一次迭代的结果:
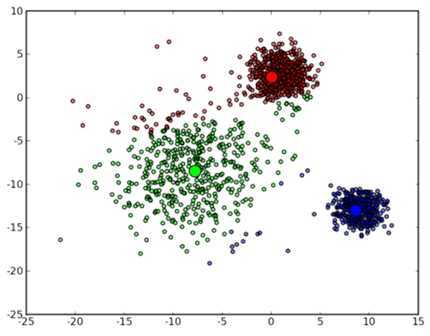
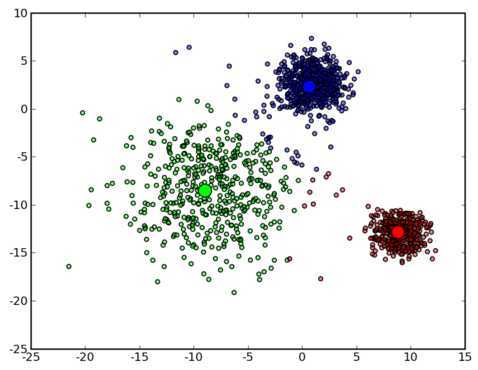
可以看到大致形状已经出来了。再经过两次迭代之后,基本上就收敛了,最终结果如下:
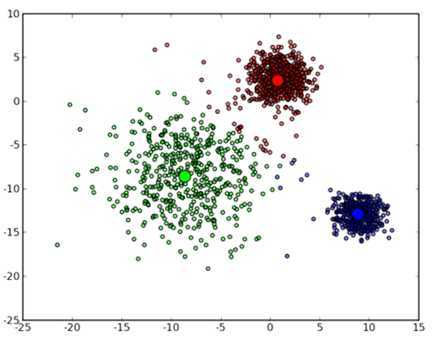
不过正如前面所说的那样 k-means 也并不是万能的,虽然许多时候都能收敛到一个比较好的结果,但是也有运气不好的时候会收敛到一个让人不满意的局部最优解,例如选用下面这几个初始中心点:
最终会收敛到这样的结果:
不得不承认这并不是很好的结果。不过其实大多数情况下 k-means 给出的结果都还是很令人满意的,算是一种简单高效应用广泛的 clustering 方法。
Update 2010.04.25: 很多人都问我要 cluster.pkl ,我干脆把它上传上来吧,其实是很容易自己生成的,点击这里下载
上一次我们了解了一个最基本的 clustering 办法 k-means ,这次要说的 k-medoids 算法,其实从名字上就可以看出来,和 k-means 肯定是非常相似的。事实也确实如此,k-medoids 可以算是 k-means 的一个变种。
k-medoids 和 k-means 不一样的地方在于中心点的选取,在 k-means 中,我们将中心点取为当前 cluster 中所有数据点的平均值:
并且我们已经证明在固定了各个数据点的 assignment 的情况下,这样选取的中心点能够把目标函数??最小化。然而在 k-medoids 中,我们将中心点的选取限制在当前 cluster 所包含的数据点的集合中。换句话说,在 k-medoids 算法中,我们将从当前 cluster 中选取这样一个点——它到其他所有(当前 cluster 中的)点的距离之和最小——作为中心点。k-means 和 k-medoids 之间的差异就类似于一个数据样本的均值 (mean) 和中位数 (median) 之间的差异:前者的取值范围可以是连续空间中的任意值,而后者只能在给样本给定的那些点里面选。那么,这样做的好处是什么呢?
一个最直接的理由就是k-means 对数据的要求太高了,它使用欧氏距离描述数据点之间的差异 (dissimilarity) ,从而可以直接通过求均值来计算中心点。这要求数据点处在一个欧氏空间之中。
然而并不是所有的数据都能满足这样的要求,对于数值类型的特征,比如身高,可以很自然地用这样的方式来处理,但是类别 (categorical) 类型的特征就不行了。举一个简单的例子,如果我现在要对犬进行聚类,并且希望直接在所有犬组成的空间中进行,k-means 就无能为力了,因为欧氏距离在这里不能用了:一只Samoyed?减去一只?Rough Collie?然后在平方一下?天知道那是什么!再加上一只?German Shepherd Dog?然后求一下平均值?根本没法算,k-means 在这里寸步难行!
在k-medoids 中,我们把原来的目标函数中的欧氏距离改为一个任意的dissimilarity measure 函数?:


Samoyed Rough Collie
最常见的方式是构造一个 dissimilarity matrix? ?来代表?,其中的元素??表示第i只狗和第?j只狗之间的差异程度,例如,两只?Samoyed?之间的差异可以设为 0 ,一只?German Shepherd Dog?和一只?Rough Collie?之间的差异是 0.7,和一只?Miniature Schnauzer?之间的差异是 1 ,等等。
?来代表?,其中的元素??表示第i只狗和第?j只狗之间的差异程度,例如,两只?Samoyed?之间的差异可以设为 0 ,一只?German Shepherd Dog?和一只?Rough Collie?之间的差异是 0.7,和一只?Miniature Schnauzer?之间的差异是 1 ,等等。
除此之外,由于中心点是在已有的数据点里面选取的,因此相对于 k-means 来说,不容易受到那些由于误差之类的原因产生的?Outlier?的影响,更加 robust 一些。
扯了这么多,还是直接来看看 k-medoids 的效果好了,由于 k-medoids 对数据的要求比 k-means 要低,所以 k-means 能处理的情况自然 k-medoids 也能处理,为了能先睹为快,我们偷一下懒,直接在上一篇文章中的 k-means 代码的基础上稍作一点修改,还用同样的例子。将代码的 45 到 47 行改成下面这样:
| |
可以看到 k-medoids 在这个例子上也能得到很好的结果:
而且,同 k-means 一样,运气不好的时候也会陷入局部最优解中:
如果仔细看上面那段代码的话,就会发现,从 k-means 变到 k-medoids ,时间复杂度陡然增加了许多:在 k-means 中只要求一个平均值??即可,而在 k-medoids 中则需要枚举每个点,并求出它到所有其他点的距离之和,复杂度为??。
看完了直观的例子,让我们再来看一个稍微实际一点的例子好了:Document Clustering ——那个永恒不变的主题,不过我们这里要做的聚类并不是针对文档的主题,而是针对文档的语言。实验数据是从?Europarl?下载的包含 Danish、German、Greek、English、Spanish、Finnish、French、Italian、Dutch、Portuguese 和 Swedish 这些语言的文本数据集。
在?N-gram-based text categorization?这篇 paper 中描述了一种计算由不同语言写成的文档的相似度的方法。一个(以字符为单位的)?N-gram?就相当于长度为 N 的一系列连续子串。例如,由 hello 产生的 3-gram 为:hel、ell 和 llo ,有时候还会在划分 N-gram 之前在开头和末尾加上空格(这里用下划线表示):_he、hel、ell、llo、lo_ 和 o__ 。按照?Zipf‘s law?:
The?nth most common word in a human language text occurs with a frequency inversely proportional to?n.
这里我们用 N-gram 来代替 word 。这样,我们从一个文档中可以得到一个 N-gram 的频率分布,按照频率排序一下,只保留频率最高的前 k 个(比如,300)N-gram,我们把叫做一个"Profile"。正常情况下,某一种语言(至少是西方国家的那些类英语的语言)写成的文档,不论主题或长短,通常得出来的 Profile 都差不多,亦即按照出现的频率排序所得到的各个 N-gram 的序号不会变化太大。这是非常好的一个性质:通常我们只要各个语言选取一篇(比较正常的,也不需要很长)文档构建出一个 Profile ,在拿到一篇未知文档的时候,只要和各个 Profile 比较一下,差异最小的那个 Profile 所对应的语言就可以认定是这篇未知文档的语言了——准确率很高,更可贵的是,所需要的训练数据非常少而且容易获得,训练出来的模型也是非常小的。
不过,我们这里且撇开分类(Classification)的问题,回到聚类(Clustering)上,按照前面的说法,在 k-medoids 聚类中,只需要定义好两个东西之间的距离(或者 dissimilarity )就可以了,对于两个 Profile ,它们之间的 dissimilarity 可以很自然地定义为对应的 N-gram 的序号之差的绝对值,在 Python 中用下面这样一个类来表示:
| ? ? ? ? ? ? ? ? ? ? ? ? |
europarl?数据集共有 11 种语言的文档,每种语言包括大约 600 多个文档。我为这七千多个文档建立了 Profile 并构造出一个 7038×7038 的 dissimilarity matrix ,最后在这上面用 k-medoids 进行聚类。构造 dissimilarity matrix 的过程很慢,在我这里花了将近 10 个小时。相比之下,k-medoids 的过程在内存允许的情况下,采用向量化的方法来做实际上还是很快的,并且通常只要数次迭代就能收敛了。实际的 k-medoids 实现可以在?mltk?中找到,今后如果有时间的话,我会陆续地把一些相关的比较通用的代码放到那里面。
最后,然我们来看看聚类的结果,由于聚类和分类不同,只是得到一些 cluster ,而并不知道这些 cluster 应该被打上什么标签,或者说,在这个问题里,包含了哪种语言的文档。由于我们的目的是衡量聚类算法的 performance ,因此直接假定这一步能实现最优的对应关系,将每个 cluster 对应到一种语言上去。一种办法是枚举所有可能的情况并选出最优解,另外,对于这样的问题,我们还可以用?Hungarian algorithm?来求解。
我们这里有 11 种语言,全排列有 11! = 39916800 种情况, 对于每一种排列,我们需要遍历一次 label list ,并数出真正的 label (语言)与聚类得出的结果相同的文档的个数,再除以总的文档个数,得到 accuracy 。假设每次遍历并求出 accuracy 只需要 1 毫秒的时间的话,总共也需要 11 个小时才能得到结果。看上去好像也不是特别恐怖,不过相比起来,用 Hungarian algorithm 的话,我们可以几乎瞬间得到结果。由于文章的篇幅已经很长了,就不在这里介绍具体的算法了,感兴趣的同学可以参考?Wikipedia?,这里我直接使用了一个现有的?Python 实现。
虽然这个实验非常折腾,不过最后的结果其实就是一个数字:accuracy ——在我这里达到了 88.97% ,证明 k-medoids 聚类和 N-gram Profile 识别语言这两种方法都是挺不错的。最后,如果有感兴趣的同学,代码可以从这里下载。需要最新版的?scipy,?munkres.py?和?mltk?以及 Python 2.6 。
在接下去说其他的聚类算法之前,让我们先插进来说一说一个有点跑题的东西:Vector Quantization?。这项技术广泛地用在信号处理以及数据压缩等领域。事实上,在 JPEG 和 MPEG-4 等多媒体压缩格式里都有 VQ 这一步。
Vector Quantization 这个名字听起来有些玄乎,其实它本身并没有这么高深。大家都知道,模拟信号是连续的值,而计算机只能处理离散的数字信号,在将模拟信号转换为数字信号的时候,我们可以用区间内的某一个值去代替着一个区间,比如,[0, 1) 上的所有值变为 0 ,[1, 2) 上的所有值变成 1 ,如此类推。其这就是一个 VQ 的过程。一个比较正式一点的定义是:VQ 是将一个向量空间中的点用其中的一个有限子集来进行编码的过程。
一个典型的例子就是图像的编码。最简单的情况,考虑一个灰度图片,0 为黑色,1 为白色,每个像素的值为 [0, 1] 上的一个实数。现在要把它编码为 256 阶的灰阶图片,一个最简单的做法就是将每一个像素值?x?映射为一个整数?floor(x*255)?。当然,原始的数据空间也并不以一定要是连续的。比如,你现在想要把压缩这个图片,每个像素只使用 4 bit (而不是原来的 8 bit)来存储,因此,要将原来的 [0, 255] 区间上的整数值用 [0, 15] 上的整数值来进行编码,一个简单的映射方案是?x*15/255?。


Rechard Stallman???????? VQ2


VQ100 VQ10
不过这样的映射方案颇有些 Naive ,虽然能减少颜色数量起到压缩的效果,但是如果原来的颜色并不是均匀分布的,那么的出来的图片质量可能并不是很好。例如,如果一个 256 阶灰阶图片完全由 0 和 13 两种颜色组成,那么通过上面的映射就会得到一个全黑的图片,因为两个颜色全都被映射到 0 了。一个更好的做法是结合聚类来选取代表性的点。
实际做法就是:将每个像素点当作一个数据,跑一下 K-means ,得到 k 个 centroids ,然后用这些 centroids 的像素值来代替对应的 cluster 里的所有点的像素值。对于彩色图片来说,也可以用同样的方法来做,例如 RGB 三色的图片,每一个像素被当作是一个 3 维向量空间中的点。
用本文开头那张?Rechard Stallman?大神的照片来做一下实验好了,VQ 2、VQ 10 和 VQ 100 三张图片分别显示聚类数目为 2 、10 和 100 时得到的结果,可以看到 VQ 100 已经和原图非常接近了。把原来的许多颜色值用 centroids 代替之后,总的颜色数量减少了,重复的颜色增加了,这种冗余正是压缩算法最喜欢的。考虑一种最简单的压缩办法:单独存储(比如 100 个)centroids 的颜色信息,然后每个像素点存储 centroid 的索引而不是颜色信息值,如果一个 RGB 颜色值需要 24 bits 来存放的话,每个(128 以内的)索引值只需要 7 bits 来存放,这样就起到了压缩的效果。
实现代码很简单,直接使用了?SciPy?提供的?kmeans?和?vq?函数,图像读写用了?Python Image Library?:
| ? ? ? ? ? ? ? ? |
当然,Vector Quantization 并不一定要用 K-means 来做,各种能用的聚类方法都可以用,只是 K-means 通常是最简单的,而且通常都够用了。
上一次我们谈到了用 k-means 进行聚类的方法,这次我们来说一下另一个很流行的算法:Gaussian Mixture Model (GMM)。事实上,GMM 和k-means 很像,不过GMM 是学习出一些概率密度函数来(所以GMM除了用在clustering上之外,还经常被用于density estimation),简单地说,k-means 的结果是每个数据点被 assign 到其中某一个cluster 了,而GMM 则给出这些数据点被 assign 到每个 cluster 的概率,又称作?soft assignment?。
得出一个概率有很多好处,因为它的信息量比简单的一个结果要多。比如,我可以把这个概率转换为一个score,表示算法对自己得出的这个结果的把握。也许我可以对同一个任务,用多个方法得到结果,最后选取"把握"最大的那个结果;另一个很常见的方法是在诸如疾病诊断之类的场所,机器对于那些很容易分辨的情况(患病或者不患病的概率很高)可以自动区分,而对于那种很难分辨的情况,比如,49% 的概率患病,51% 的概率正常,如果仅仅简单地使用 50% 的阈值将患者诊断为"正常"的话,风险是非常大的,因此,在机器对自己的结果把握很小的情况下,会"拒绝发表评论",而把这个任务留给有经验的医生去解决。
废话说了一堆,不过,在回到 GMM 之前,我们再稍微扯几句。我们知道,不管是机器还是人,学习的过程都可以看作是一种"归纳"的过程,在归纳的时候你需要有一些假设的前提条件,例如,当你被告知水里游的那个家伙是鱼之后,你使用"在同样的地方生活的是同一种东西"这类似的假设,归纳出"在水里游的都是鱼"这样一个结论。当然这个过程是完全"本能"的,如果不仔细去想,你也不会了解自己是怎样"认识鱼"的。另一个值得注意的地方是这样的假设并不总是完全正确的,甚至可以说总是会有这样那样的缺陷的,因此你有可能会把虾、龟、甚至是潜水员当做鱼。也许你觉得可以通过修改前提假设来解决这个问题,例如,基于"生活在同样的地方并且穿着同样衣服的是同一种东西"这个假设,你得出结论:在水里有并且身上长有鳞片的是鱼。可是这样还是有问题,因为有些没有长鳞片的鱼现在又被你排除在外了。
在这个问题上,机器学习面临着和人一样的问题,在机器学习中,一个学习算法也会有一个前提假设,这里被称作"归纳偏执 (bias)"(bias 这个英文词在机器学习和统计里还有其他许多的意思)。例如线性回归,目的是要找一个函数尽可能好地拟合给定的数据点,它的归纳偏执就是"满足要求的函数必须是线性函数"。一个没有归纳偏执的学习算法从某种意义上来说毫无用处,就像一个完全没有归纳能力的人一样,在第一次看到鱼的时候有人告诉他那是鱼,下次看到另一条鱼了,他并不知道那也是鱼,因为两条鱼总有一些地方不一样的,或者就算是同一条鱼,在河里不同的地方看到,或者只是看到的时间不一样,也会被他认为是不同的,因为他无法归纳,无法提取主要矛盾、忽略次要因素,只好要求所有的条件都完全一样──然而哲学家已经告诉过我们了:世界上不会有任何样东西是完全一样的,所以这个人即使是有无比强悍的记忆力,也绝学不到任何一点知识。
这个问题在机器学习中称作"过拟合 (Overfitting)",例如前面的回归的问题,如果去掉"线性函数"这个归纳偏执,因为对于 N 个点,我们总是可以构造一个 N-1 次多项式函数,让它完美地穿过所有的这 N 个点,或者如果我用任何大于 N-1 次的多项式函数的话,我甚至可以构造出无穷多个满足条件的函数出来。如果假定特定领域里的问题所给定的数据个数总是有个上限的话,我可以取一个足够大的 N ,从而得到一个(或者无穷多个)"超级函数",能够 fit 这个领域内所有的问题。然而这个(或者这无穷多个)"超级函数"有用吗?只要我们注意到学习的目的(通常)不是解释现有的事物,而是从中归纳出知识,并能应用到新的事物上,结果就显而易见了。
没有归纳偏执或者归纳偏执太宽泛会导致 Overfitting ,然而另一个极端──限制过大的归纳偏执也是有问题的:如果数据本身并不是线性的,强行用线性函数去做回归通常并不能得到好结果。难点正在于在这之间寻找一个平衡点。不过人在这里相对于(现在的)机器来说有一个很大的优势:人通常不会孤立地用某一个独立的系统和模型去处理问题,一个人每天都会从各个来源获取大量的信息,并且通过各种手段进行整合处理,归纳所得的所有知识最终得以统一地存储起来,并能有机地组合起来去解决特定的问题。这里的"有机"这个词很有意思,搞理论的人总能提出各种各样的模型,并且这些模型都有严格的理论基础保证能达到期望的目的,然而绝大多数模型都会有那么一些"参数"(例如 K-means 中的 k),通常没有理论来说明参数取哪个值更好,而模型实际的效果却通常和参数是否取到最优值有很大的关系,我觉得,在这里"有机"不妨看作是所有模型的参数已经自动地取到了最优值。另外,虽然进展不大,但是人们也一直都期望在计算机领域也建立起一个统一的知识系统(例如语意网就是这样一个尝试)。
废话终于说完了,回到 GMM 。按照我们前面的讨论,作为一个流行的算法,GMM 肯定有它自己的一个相当体面的归纳偏执了。其实它的假设非常简单,顾名思义,Gaussian Mixture Model ,就是假设数据服从 Mixture Gaussian Distribution ,换句话说,数据可以看作是从数个 Gaussian Distribution 中生成出来的。实际上,我们在?K-means?和?K-medoids?两篇文章中用到的那个例子就是由三个 Gaussian 分布从随机选取出来的。实际上,从中心极限定理可以看出,Gaussian 分布(也叫做正态 (Normal) 分布)这个假设其实是比较合理的,除此之外,Gaussian 分布在计算上也有一些很好的性质,所以,虽然我们可以用不同的分布来随意地构造 XX Mixture Model ,但是还是 GMM 最为流行。另外,Mixture Model 本身其实也是可以变得任意复杂的,通过增加 Model 的个数,我们可以任意地逼近任何连续的概率密分布。
每个 GMM 由?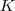 ?个 Gaussian 分布组成,每个 Gaussian 称为一个"Component",这些 Component 线性加成在一起就组成了 GMM 的概率密度函数:
?个 Gaussian 分布组成,每个 Gaussian 称为一个"Component",这些 Component 线性加成在一起就组成了 GMM 的概率密度函数:
根据上面的式子,如果我们要从GMM 的分布中随机地取一个点的话,实际上可以分为两步:首先随机地在这?K个 Component 之中选一个,每个 Component 被选中的概率实际上就是它的系数??,选中了 Component 之后,再单独地考虑从这个 Component 的分布中选取一个点就可以了──这里已经回到了普通的 Gaussian 分布,转化为了已知的问题。
那么如何用 GMM 来做 clustering 呢?其实很简单,现在我们有了数据,假定它们是由 GMM 生成出来的,那么我们只要根据数据推出 GMM 的概率分布来就可以了,然后 GMM 的?K个 Component 实际上就对应了?K?个 cluster 了。根据数据来推算概率密度通常被称作 density estimation ,特别地,当我们在已知(或假定)了概率密度函数的形式,而要估计其中的参数的过程被称作"参数估计"。
现在假设我们有?N?个数据点,并假设它们服从某个分布(记作??),现在要确定里面的一些参数的值,例如,在 GMM 中,我们就需要确定?、?和??这些参数。 我们的想法是,找到这样一组参数,它所确定的概率分布生成这些给定的数据点的概率最大,而这个概率实际上就等于,我们把这个乘积称作似然函数 (Likelihood Function)。通常单个点的概率都很小,许多很小的数字相乘起来在计算机里很容易造成浮点数下溢,因此我们通常会对其取对数,把乘积变成加和?,得到 log-likelihood function 。接下来我们只要将这个函数最大化(通常的做法是求导并令导数等于零,然后解方程),亦即找到这样一组参数值,它让似然函数取得最大值,我们就认为这是最合适的参数,这样就完成了参数估计的过程。
下面让我们来看一看 GMM 的 log-likelihood function:
由于在对数函数里面又有加和,我们没法直接用求导解方程的办法直接求得最大值。为了解决这个问题,我们采取之前从 GMM 中随机选点的办法:分成两步,实际上也就类似于?K-means?的两步。
估计数据由每个 Component 生成的概率(并不是每个 Component 被选中的概率):对于每个数据??来说,它由第?k?个 Component 生成的概率为
由于式子里的??和??也是需要我们估计的值,我们采用迭代法,在计算?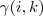 ?的时候我们假定??和?均已知,我们将取上一次迭代所得的值(或者初始值)。
?的时候我们假定??和?均已知,我们将取上一次迭代所得的值(或者初始值)。
估计每个 Component 的参数:现在我们假设上一步中得到的??就是正确的"数据?由 Component?k?生成的概率",亦可以当做该 Component 在生成这个数据上所做的贡献,或者说,我们可以看作??这个值其中有?这部分是由 Component?k?所生成的。集中考虑所有的数据点,现在实际上可以看作 Component 生成了??这些点。由于每个 Component 都是一个标准的 Gaussian 分布,可以很容易分布求出最大似然所对应的参数值:
其中,并且??也顺理成章地可以估计为?。
重复迭代前面两步,直到似然函数的值收敛为止。
当然,上面给出的只是比较"直观"的解释,想看严格的推到过程的话,可以参考?Pattern Recognition and Machine Learning?这本书的第九章。有了实际的步骤,再实现起来就很简单了。Matlab 代码如下:
(Update 2012.07.03:如果你直接把下面的代码拿去运行了,碰到 covariance 矩阵 singular 的情况,可以参见这篇文章。)
| ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? |
函数返回的?Px?是一个??的矩阵,对于每一个??,我们只要取该矩阵第?i?行中最大的那个概率值所对应的那个 Component 为?所属的 cluster 就可以实现一个完整的聚类方法了。对于最开始的那个例子,GMM 给出的结果如下:
?

相对于之前 K-means 给出的结果,这里的结果更好一些,左下角的比较稀疏的那个 cluster 有一些点跑得比较远了。当然,因为这个问题原本就是完全有 Mixture Gaussian Distribution 生成的数据,GMM (如果能求得全局最优解的话)显然是可以对这个问题做到的最好的建模。
另外,从上面的分析中我们可以看到 GMM 和 K-means 的迭代求解法其实非常相似(都可以追溯到?EM 算法,下一次会详细介绍),因此也有和 K-means 同样的问题──并不能保证总是能取到全局最优,如果运气比较差,取到不好的初始值,就有可能得到很差的结果。对于 K-means 的情况,我们通常是重复一定次数然后取最好的结果,不过 GMM 每一次迭代的计算量比 K-means 要大许多,一个更流行的做法是先用 K-means (已经重复并取最优值了)得到一个粗略的结果,然后将其作为初值(只要将 K-means 所得的 centroids 传入?gmm?函数即可),再用 GMM 进行细致迭代。
如我们最开始所讨论的,GMM 所得的结果(Px)不仅仅是数据点的 label ,而包含了数据点标记为每个 label 的概率,很多时候这实际上是非常有用的信息。最后,需要指出的是,GMM 本身只是一个模型,我们这里给出的迭代的办法并不是唯一的求解方法。感兴趣的同学可以自行查找相关资料。
我之前写过一篇介绍 Gaussian Mixture Model (GMM) 的文章,并在文章里贴了一段 GMM实现的 Matlab 示例代码,然后就不断地有人来问我关于那段代码的问题,问得最多的就是大家经常发现在跑那段代码的时候估计出来的 Covariance Matrix 是 singular 的,所以在第 96 行求逆的时候会挂掉。这是今天要介绍的主要话题,我会讲得罗嗦一点,把关于那篇文章我被问到的一些其他问题也都提到一下,不过,在步入正题之前,不妨先来小小地闲扯一下。
我自己以前就非常在痛恨书里看到伪代码,又不能编译运行,还搞得像模像样的像代码一般,所以我自己在写 blog 的时候就尝试给出直接可运行的代码,但是现在我渐渐理解为什么书的作者喜欢用伪代码而不是可运行的代码了(除非是专门讲某编程语言的书)。原因很简单:示例用的代码和真实项目中的代码往往是差别很大的,直接给出可运行的示例代码,读者就会直接拿去运行(因为包括我在内的大部分人都是很懒的,不是说"懒"是程序员的天性么?),而往往示例代码为了结构清晰并突出算法的主要脉络,对很多琐碎情况都没有处理,都使用最直接的操作,没有做任何优化(例如我的那个 GMM 示例代码里直接用 matlab 的inv?函数来求 Covariance 矩阵的逆),等等,因此直接运行这样的代码——特别是在实际项目中使用,是非常不明智的。
可是那又为什么不直接给出实际项目级别的代码呢?第一个原因当然是工作量,我想程序员都知道从一个算法到一个健壮、高效的系统之间有多长的路要走,而且很多时候都需要根据不同的项目环境和需要做不同的修改。所以当一个人实际上是在介绍算法的时候让他去弄一套工业级别的代码出来作为示例,实在是太费力了。当然更重要的原因是:工业级别的代码通常经过大量的优化并充斥着大量错误处理、以及为了处理其他地方的 bug 或者整个系统混乱的 API 等而做的各种 workaround 等等……也许根本就看不懂,基本完全失去了示例的作用。所以,伪代码就成为了唯一的选择。
罗嗦了半天,现在我们回到正题,那篇文章讲的是 GMM 的学习,所以关于 GMM 的问题可以直接参考那篇文章,出问题的地方仅在于 GMM 的每个 component (Gaussian Distribution) 的参数估计上,特别地,在于 Covariance Matrix 的估计上。所以,我们今天主要来探讨 Gaussian 分布的协方差矩阵的估计问题。首先来看一下多维 Gaussian 分布的概率密度函数:
eq: 1 ?
参数估计的过程是给定一堆数据点?,并假设他们是由某个真实的 Gaussian 分布独立地 (IID) 采样出来的,然后根据这堆点来估计该 Gaussian 分布的参数。对于一个多维 Gaussian 分布来说,需要估计的就是均值??和协方差矩阵??两"个"参数——用引号是因为?和??都不是标量,假设我们的数据是??维的,那么??实际上是??个参数,而作为一个?的矩阵,?则是由??个参数组成。根据协方差矩阵的对称性,我们可以进一步把??所包含的参数个数降低为??个。
值得一提的是,我们从参数估计的角度并不能一下子断定??就是对称的,需要稍加说明(这实际上是《Pattern Recognition and Machine Learning》书里的习题 2.17)。记?,假设我们的??不是对称的,那么我们把他写成一个对称矩阵和一个反对称阵之和(任何一个矩阵都可以这样表示):
将起代入式?(eq: 1)?的指数部分中,注意到对于反对称部分(书写方便起见我们暂时另?):
所以指数部分只剩下对称阵??了。也就是说,如果我们找出一个符合条件的非对称阵?,用它的对称部分??代替也是可以的,所以我们就可以"不失一般性"地假设??是对称的(因为对称阵的逆矩阵也是对称的)。注意我们不用考虑式?(eq: 1)?指数前面的系数(里的那个??的行列式),因为前面的系数是起 normalization 作用的,使得全概率积分等于 1,指数部分变动之后只要重新 normalize 一下即可(可以想像一下,如果??真的是非对称的,那么 normalization 系数那里应该也不会是??那么简单的形式吧?有兴趣的同学可以自己研究一下)。
好,现在回到参数估计上。对于给定的数据?,将其代入式?(eq: 1)?中可以得到其对应的 likelihood?,注意这个并不是??的概率。和离散概率分布不同的是,这里谈论单个数据点的概率是没有多大意义的,因为单点集的测度为零,它的概率也总是为零。所以,这里的 likelihood 出现大于 1 的值也是很正常的——这也是我经常被问到的一个问题。虽然不是概率值,但是我想从 likelihood 或者概率密度的角度去理解,应该也是可以得到直观的认识的。
参数估计常用的方法是最大似然估计(Maximum Likelihood Estimation, MLE)。就是将所有给定数据点的似然全部乘起来,得到一个关于参数(这里是??和?)的函数,然后最大化该函数,所对应的参数值就是最大似然估计的值。通常为了避免数值下溢,会使用单调函数??将相乘变成相加在计算。
对于最大似然估计,我的理解是寻找最可能生成给定的数据的模型参数。这对于离散型的分布来说是比较好解释的,因为此时一点的似然实际上就是该点的概率,不过对于像 Gaussian 这样的针对连续数据的分布来说,就不是那么自然了。不过我们其实有另一种解释,这是我在Coursera?的?Probabilistic Graphical Model?课上看到的:
对于一个真实的分布?,假设我们得到了它的一个估计?,那么如何来衡量这个估计的好坏呢?比较两个概率分布的一个常用的办法是用?KL-Divergence,又叫 Relative Entropy(以及其他若干外号),定义为:
不过由于我们通常是不知道真实的??的,所以这个值也没法直接算,于是我们把它简单地变形一下:
其中蓝色部分是分布??自己的?Entropy,由于我们比较不同的模型估计好坏的时候这个量是不变的,所以我们可以忽略它,只考虑红色部分。为了使得 KL-Divergence 最小,我们需要将红色部分(注意不包括前面的负号)最大化。不过由于仍然依赖于真实分布?,所以红色部分还是不能直接计算,但是这是一个我们熟悉的形式:某个函数关于真实分布的期望值,而我们手里面又有从真实分布里 IID 采样出来的??个数据点,所以可以直接使用标准的近似方法:用数据的经验分布期望来代替原始的期望:
于是我们很自然地就得到了最大似然估计的目标函数。这也从另一个角度解释了最大似然估计的合理性:同时对离散型和连续型的数据都适用。
接下来我们再回到 Gaussian 分布,将 Gaussian 的概率密度函数?(eq: 1)?代入最大似然估计的目标函数,可以得到:
eq: 2 ?
由于本文主要讨论协方差矩阵的估计,我们这里就不管均值矩阵??的估计了,方法上是类似的,下面式子中的??将表示已知的或者同样通过最大似然估计计算出来的均值。为了找到使得上式最大化的协方差矩阵,我们将它对于??求导:
另导数等于零,即可得到最优解:
eq: 3 ?
于是??就是协方差矩阵的最大似然估计。到这里问题就出来了,再回到 Gaussian 分布的概率密度函数?(eq: 1),如果把??带进去的话,可以看到是需要对它进行求逆的,这也是我被问得最多的问题:由于??singular?了,导致求逆失败,于是代码也就运行失败了。
从?(eq: 3)?可以大致看到什么时候??会 singular:由于该式的形式,可以知道??的?rank?最大不会超过?,所以如果?,也就是数据的维度大于数据点的个数的话,得到的估计值??肯定是不满秩的,于是就 singular 了。当然,即使不是这么极端的情况,如果??比较小或者?比较大的时候,仍然会有很大的几率出现不满秩的。
这个问题还可以从更抽象一点的角度来看。我们之前计算过估计 Covariance 矩阵实际上是要估计??这么多个参数,如果??比较大的话,这个数目也会变得非常多,也就意味着模型的复杂度很大,对于很复杂的模型,如果训练数据的量不够多,则无法确定一个唯一的最优模型出来,这是机器学习中非常常见的一个问题,通常称为?overfitting。解决 overfitting 问题的方法有不少,最直接的就是用更多的训练数据(也就是增大?),当然,有时候训练数据只有那么多,于是就只好从反面入手,降低模型复杂度。
在降低模型复杂度方面最常用的方法大概是正则化了,而正则化最简单的例子也许是?Ridge Regression?了,通过在目标函数后面添加一项关于参数的?-norm 的项来对模型参数进行限制;此外也有诸如 LASSO 之类的使用?-norm 来实现稀疏性的正则化,这个我们在之前也曾经简单介绍过。
在介绍估计 Covariance 的正则化方法之前,我们首先来看一种更直接的方法,直接限制模型的复杂度:特别地,对于我们这里 Gaussian 的例子,比如,我们可以要求协方差矩阵是一个对角阵,这样一来,对于协方差矩阵,我们需要估计的参数个数就变成了??个对角元素,而不是原来的??个。大大减少参数个数的情况下,overfitting 的可能性也极大地降低了。
注意 Covariance Matrix 的非对角线上的元素是对应了某两个维度之间的相关性 (Correlation) 的,为零就表示不相关。而对于 Gaussian 分布来说,不相关和独立 (Independence) 是等价的,所以说在我们的假设下,向量??的各个维度之间是相互独立的,换句话说,他们的联合概率密度可以分解为每个维度各自的概率密度(仍然是 Gaussian 分布)的乘积,这个特性使得我们可以很方便地对协方差矩阵进行估计:只有每个维度上当做一维的 Gaussian 分布各自独立地进行参数估计就可以了。注意这个时候除非某个维度上的坐标全部是相等的,否则 Covariance 矩阵对角线上的元素都能保证大于零,也就不会出现 singular 的情况了。
那么直观上将协方差矩阵限制为对角阵会产生什么样的模型呢?我们不妨看一个二维的简单例子。下面的代码利用了?scikit-learn?里的 GMM 模块来估计一个单个 component 的 Gaussian,它在选项里已经提供了 Covariance 的限制,这里我们除了原始的?full?和对角阵的?diag?之外,还给出一个?spherical,它假定协方差矩阵是??这样的形式,也就是说,不仅是对角阵,而且所有对角元素都相等。下面的代码把三种情况下 fit 出来的模型画出来,在图?(fig: 1)中可以看到。
import
numpy
as
np
import
pylab
as
pl
from
sklearn
import mixture
n_samples =
300
c_types = [‘full‘, ‘diag‘, ‘spherical‘]
np.random.seed(0)
C = np.array([[0., -0.7], [3.5, 1.7]])
X_train = np.dot(np.random.randn(n_samples, 2), C)
pl.figure(dpi=100,figsize=(3,3))
pl.scatter(X_train[:, 0], X_train[:, 1], .8)
pl.axis(‘tight‘)
pl.savefig(‘GaussianFit-data.svg‘)
pl.close()
for c_type in c_types:
clf = mixture.GMM(n_components=1, covariance_type=c_type)
clf.fit(X_train)
x = np.linspace(-15.0, 20.0, num=200)
y = np.linspace(-10.0, 10.0, num=200)
X, Y = np.meshgrid(x, y)
XX = np.c_[X.ravel(), Y.ravel()]
Z = np.log(-clf.eval(XX)[0])
Z = Z.reshape(X.shape)
pl.figure(dpi=100,figsize=(3,3))
CS = pl.contour(X, Y, Z)
pl.scatter(X_train[:, 0], X_train[:, 1], .8)
pl.axis(‘tight‘)
pl.savefig(‘GaussianFit-%s.svg‘
% c_type)
pl.close()
fig: 1 ?
Training samples.


Spherical Covariance.


Diagonal Covariance.


Full Covariance.
可以看到,从直观上来讲:spherical Covariance 的时候就是各个维度上的方差都是一样的,所以形成的等高线在二维上是正圆。而 diagonal Covariance 则可以 capture 更多的特征,因为它允许形成椭圆的等高线,但是它的限制是这个椭圆是不能旋转的,永远是正放的。Full Covariance 是 fit 最好的,但是前提是数据量足够来估计这么多的参数,虽然对于 2 维这样的低维度通常并不是问题,但是维度高了之后就经常会变得很困难。
一般来说,使用对角线的协方差矩阵是一种不错的降低模型复杂度的方式,在 scikit-learn 的 GMM 模块中这甚至是默认的。不过如果有时候你觉得这样的限制实在是和你的真实数据想去甚远的话,也有另外的处理方式。最简单的处理办法是在求出了 Covariance 矩阵的估计值之后直接加上一个?,也就是在对角线上统一加上一个很小的正数?,通常称作正则化系数。例如,Matlab 自带的统计工具箱里的?gmdistribution.fit?函数就有一个可选参数叫做Regularize,就是我们这里说的?。为什么加上??之后就不会 singular 了呢?首先??本身至少肯定是半正定的,所以?:
所以这样之后肯定就是正定的了。此外在 scikit-learn 的 GMM 学习的函数中也有一个类似的参数?min_covar,它的文档里说的是"Floor on the diagonal of the covariance matrix to prevent overfitting. Defaults to 1e-3."感觉好像是如果原来的 Covariance 矩阵对角线元素小于该参数的时候就将它设置为该参数,不过这样的做法是不是一定产生正定的协方差矩阵似乎就不像上面那种那样直观可以得出结论了。
不过这样的做法虽然能够解决 singular 的问题,但是总让人有些难以信服:你莫名其妙地在我的协方差矩阵上加上了一些东西,这到底是什么意思呢?最简单的解释可以从式?(eq: 1)?那里协方差矩阵你的地方来看,对于矩阵求逆或者解方程的时候出现 singular 的情况,加上一个?也算是数值上的一种标准处理方式,叫做?Tikhonov Regularization,Ridge Regression 本身也可以通过这个角度来解释。从数值计算的角度来说,这样处理能够增加数值稳定性,直观地来讲,稳定性是指??元素的微小数值变化,不会使得求逆(或者解方程之类的)之后的解产生巨大的数值变化,如果??是 singular 的话,通常就不具有这样的稳定性了。此外,随着 regularization coefficient??逐渐趋向于零,对应的解也会逐渐趋向于真实的解。
如果这个解释还不能令出信服的话,我们还可以从 Bayes 推断的角度来看这个问题。不过这本身是一个很大的话题,我在这里只能简略地讲一个思路,想了解更多的同学可以参考《Pattern Recognition and Machine Learning》第二章或者其他相关的书籍。
总的来说,我们这里要通过?Maximum a posteriori (MAP)?Estimation 来对协方差矩阵进行估计。做法是首先为??定义一个先验分布 (prior)?,然后对于数据?,我们根据 Bayes 公式,有
eq: 4 ?
其中等式左边称作后验 (posterior),右边红色部分是似然 (likelihood),蓝色部分是用于将概率分布 normalize 到全概率为 1 的,通常并不需要直接通过??去计算,而只要对求出的概率分布进行归一化就可以了。MAP 的思路是将 posterior 最大的那个??作为最佳估计值,直观上很好理解,就是给定了数据点之后"最可能的"那个?。而计算上我们是通过?(eq: 4)?来实现的,因为分母是与??无关的,所以在计算的时候(因为只要比较相对大小嘛)忽略掉,这样一来,就可以看到,其实 MAP 方法和刚才我们用的最大似然 (MLE) 方法唯一的区别就在于前面乘了一个先验分布。
虽然从最终的计算公式上来看差别很细微,但是 MAP 却有很多好处。和本文最相关的当然是先验的引入,这在很大程度上和我们平时用正则化的目的是一样的:加入我们自己对模型的先验知识或者假设或者要求,这样可以避免在数据不够的时候产生 overfitting 。此外还有另一个值得一提的好玩特性就是 MAP 可以在线计算:注意到后验分布本身也是关于??的一个合法分布,所以在我们计算出后验之后,如果又得到了新的训练数据,那么我们可以将之前算出来的后验又作为先验,代入新的一轮的计算,这样可以一直不断地重复下去。:D
不过,虽然 MAP 框架看上去很美,但是在 general 的情况下计算却可能会变得很复杂,因此,虽然理论上来说,我们可以把任何先验知识通过先验分布的方式加入到模型中来,但是在实际中先验的选择往往是根据"哪个更好计算"而不是根据"哪个更合理"的准则来做出的。基于这个准则,有了?Conjugate Prior?的概念,简单地来说,如果一个 prior 和 likelihood 相乘再归一化之后得到的 posterior 形式上是和 prior 一样的话,那么它就是该 likelihood 的一个 conjugate prior。
选择 conjugate prior 的好处是显而易见的,由于先验和后验的形式是一样的,都是某一类型的分布,只是参数的值发生了变化,所以说可以看成是在观察到数据之后对模型参数进行修正(注意这里的模型是关于??的,而??本身又是关于数据的模型——一个 Gaussian 分布——的参数,不要搞混淆了),并且这种修正有时候可以得到非常直观的解释,不过我在这里就不细说了。此外,由于形式没有发生变化,所以在使用在线计算的时候,每一次观察到新的数据所使用的计算公式都还是一样的。
回到我们的问题,关于 Gaussian 分布的协方差矩阵的 Conjugate Prior 是一个叫做?Inverse Wishart Distribution?的家伙,它的概率密度函数是这个样子的:
其中??和参数??都是??的正定矩阵,而??则是?Multivariate Gamma Function。是不是看上去有点恐怖?^_^bbbb我也觉得有点恐怖……接下来让我们来看 MAP,首先将这个 prior 乘以我们的 likelihood,和 MLE 一样的,我们在??空间中计算,所以对整个式子取对数,然后丢掉和??无关的常数项(因为是关于??最大化嘛),得到下面的式子:
可以看到现在已经变得不是那么复杂了,形式上和我们之前的?(eq: 2)?是一样的,只是多了红色的一项,接下来对??进行求导并令导数等于零,类似地得到如下方程:
于是得到最优解为
这里??是之前的最大似然估计。接下来如果我们将我们 prior 中的参数??选为?,就可以得到 MAP 估计为:
就得到了我们刚才的正则化的形式。有一点细微的区别就是这里前面多了一个全局的缩放系数,不过这样的系数是没有关系的,可以直接丢掉,因为它总也会在最终归一化的时候被 Gaussian 分布前面的系数给抵消掉。这样一来,我们就得到了这种正则化方法的一个看起来比较靠谱的解释:实际上就是我们先验地假设协方差矩阵满足一个均值为 spherical 形状(所有对角元素都相等的一个对角阵)的 Inverse Wishart 分布(关于 Inverse Wishart 分布的均值的公式可以参见?wikipedia),然后通过数据来对这个先验进行修正得到后验概率,并从后验中取了概率(密度)最大的那个??作为最终的估计。
这种方式比暴力地直接强制要求协方差矩阵具有 diagonal 或者 spherical 形式要温柔得多,并且随着训练数据的增多,先验的影响也会逐渐减小,从而趋向于得到真实的参数,而强制结构限制的结果则是不论你最后通过什么手段搞到了多少数据,如果真实模型本身不是 diagonal 或者 spherical 的话,那么你的参数估计的 bias 将永远都无法被消除,这个我们已经在图?(fig: 1)中直观地看到了。当然,在训练数据本身就不足的情况下,两种选择谁好谁好就没有定论了,不要忘了,强制结构限制的计算复杂度要小很多。:)
最后,对于不想看长篇大论的人,我做一下简单的总结:如果碰到估计 Gaussian 的协方差矩阵时结果 singular 的情况,解决方法一般有两种:
然后再补充几点不知道该放在哪里的注意事项:
Expectation Maximization (EM)?是一种以迭代的方式来解决一类特殊最大似然 (Maximum Likelihood) 问题的方法,这类问题通常是无法直接求得最优解,但是如果引入隐含变量,在已知隐含变量的值的情况下,就可以转化为简单的情况,直接求得最大似然解。
我们会看到,上一次说到的?Gaussian Mixture Model?的迭代求解方法可以算是 EM 算法最典型的应用,而最开始说的?K-means?其实也可以看作是 Gaussian Mixture Model 的一个变种(固定所有的?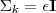 ?,并令?
?,并令? ?即可)。然而 EM 实际上是一种通用的算法(或者说是框架),可以用来解决很多类似的问题,我们最后将以一个中文分词的例子来说明这一点。
?即可)。然而 EM 实际上是一种通用的算法(或者说是框架),可以用来解决很多类似的问题,我们最后将以一个中文分词的例子来说明这一点。
为了避免问题变得太抽象,我们还是先从上一次的 Gaussian Mixture Model 说起吧。回顾一下我们之前要解决的问题:求以下 Log-likelihood function 的最大值:
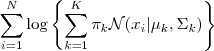
但是由于在?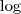 ?函数里面又有加和,没法直接求。考虑一下 GMM 生成 sample 的过程:先选一个 Component ,然后再从这个 Component 所对应的那个普通的 Gaussian Distribution 里进行 sample 。我们可以很自然地引入一个隐含变量?
?函数里面又有加和,没法直接求。考虑一下 GMM 生成 sample 的过程:先选一个 Component ,然后再从这个 Component 所对应的那个普通的 Gaussian Distribution 里进行 sample 。我们可以很自然地引入一个隐含变量? ?,这是一个?
?,这是一个?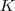 ?维向量,如果第?
?维向量,如果第?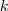 ?个 Component 被选中了,那么我们讲其第?
?个 Component 被选中了,那么我们讲其第?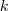 ?个元素置为 1 ,其余的全为 0 。那么,再来看看,如果除了之前的 sample?
?个元素置为 1 ,其余的全为 0 。那么,再来看看,如果除了之前的 sample?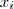 ?的值之外,我们同时还知道了每个?
?的值之外,我们同时还知道了每个?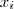 ?所对应的隐含变量?
?所对应的隐含变量?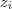 ?的值,情况又会变成怎么样呢?
?的值,情况又会变成怎么样呢?
因为我们同时观察到了? ?和?
?和? ?,所以我们现在要最大化的似然函数也变为?
?,所以我们现在要最大化的似然函数也变为?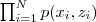 ?。注意到?
?。注意到?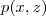 ?可以表示为:
?可以表示为:
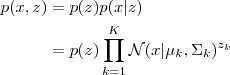
而? ?的概率,当?
?的概率,当? ?的第?
?的第?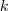 ?个元素为 1 的时候,亦即第?
?个元素为 1 的时候,亦即第?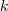 ?个 Component 被选中的时候,这个概率为?
?个 Component 被选中的时候,这个概率为?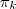 ?,统一地写出来就是:
?,统一地写出来就是:
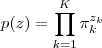
带入上面个式子,我们得到?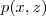 ?的概率是一个大的乘积式(对比之前?
?的概率是一个大的乘积式(对比之前?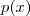 ?是一个和式)。再替换到最开始的那个 Log-likelihood function 中,得到新的同时关于 sample?
?是一个和式)。再替换到最开始的那个 Log-likelihood function 中,得到新的同时关于 sample? ?和隐含变量?
?和隐含变量? ?的 Log-likelihood:
?的 Log-likelihood:
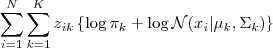
情况瞬间逆转,现在?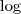 ?和求和符号换了个位置,直接作用在普通的高斯分布上了,一下子就变成了可以直接求解的问题。不过,事情之所以能发展得如此顺利,完全依赖于一个我们伪造的假设:隐含变量的值已知。然而实际上我们并不知道这个值。问题的结症在这里了,如果我们有了这个值,所有的问题都迎刃而解了。回想一下,在类似的地方,我们是如何处理这样的情况的呢?一个很类似的地方就是(比如,在数据挖掘中)处理缺失数据的情况,一般有几种办法:
?和求和符号换了个位置,直接作用在普通的高斯分布上了,一下子就变成了可以直接求解的问题。不过,事情之所以能发展得如此顺利,完全依赖于一个我们伪造的假设:隐含变量的值已知。然而实际上我们并不知道这个值。问题的结症在这里了,如果我们有了这个值,所有的问题都迎刃而解了。回想一下,在类似的地方,我们是如何处理这样的情况的呢?一个很类似的地方就是(比如,在数据挖掘中)处理缺失数据的情况,一般有几种办法:
这里我们采取一种类似于平均值的办法:取期望。因为这里我们至少有 sample? ?的值,因此我们可以把这个信息利用起来,针对后验概率?
?的值,因此我们可以把这个信息利用起来,针对后验概率?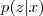 ?来取期望。前面说过,
?来取期望。前面说过, ?的每一个元素只有 0 和 1 两种取值,因此按照期望的公式写出来就是:
?的每一个元素只有 0 和 1 两种取值,因此按照期望的公式写出来就是:
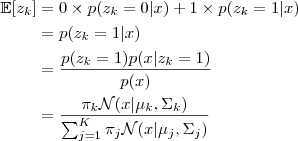
中间用贝叶斯定理变了一下形,最后得到的式子正是我们在漫谈 GMM?中定义的?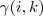 ?。因此,对于上面那个可以直接求解的 Log-likelihood function 中的?
?。因此,对于上面那个可以直接求解的 Log-likelihood function 中的?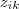 ?,我们只要用其期望?
?,我们只要用其期望?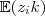 亦即?
亦即?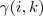 ?代替即可。
?代替即可。
到这里为止,看起来是一个非常完美的方法,不过仔细一看,发现还有一个漏洞:之前的 Log-likelihood function 可以直接求最大值是建立在?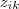 ?是已知情况下,现在虽然我们用?
?是已知情况下,现在虽然我们用?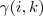 ?来代替了?
?来代替了?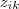 ,但是实际上?
,但是实际上?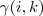 ?是一个反过来以非常复杂的关系依赖所要求参数的一个式子,而不是一个"已知的数值"。解决这个问题的办法就是迭代。
?是一个反过来以非常复杂的关系依赖所要求参数的一个式子,而不是一个"已知的数值"。解决这个问题的办法就是迭代。
到此为止,我们就勾勒出了整个 EM 算法的结构,同时也把上一次所讲的求解 GMM 的方法又推导了一遍。EM 名字也来源于此:
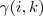 ;
;
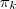 、
、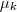 和?
和?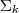 ?。
?。
如果你还没有厌烦这些公式的话,让我们不妨再稍微花一点时间,从偏理论一点的角度来简略地证明一下 EM 这个迭代的过程每一步都会对结果有所改进,除非已经达到了一个(至少是局部的)最优解。
现在我们的讨论将不局限于 GMM ,并使用一些稍微紧凑一点的符号。用? ?表示所有的 sample ,同时用?
?表示所有的 sample ,同时用?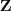 ?表示所有对应的隐含变量。我们的问题是要通过最大似然的方法估计出?
?表示所有对应的隐含变量。我们的问题是要通过最大似然的方法估计出?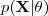 ?中的参数?
?中的参数? ?。在这里我们假设这个问题很困难,但是要优化?
?。在这里我们假设这个问题很困难,但是要优化?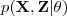 ?却很容易。这就是 EM 算法能够解决的那一类问题。
?却很容易。这就是 EM 算法能够解决的那一类问题。
现在我们引入一个关于隐含变量的分布?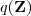 。注意到对于任意的?
。注意到对于任意的?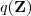 ?,我们都可以对 Log-likelihood Function 作如下分解:
?,我们都可以对 Log-likelihood Function 作如下分解:
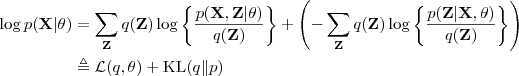
其中?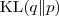 ?是分布?
?是分布?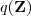 ?和?
?和?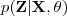 ?之间的?Kullback-Leibler divergence?。由于 Kullback-Leibler divergence 是非负的,并且只有当两个分布完全相同的时候才会取到 0 。因此我们可以得到关系?
?之间的?Kullback-Leibler divergence?。由于 Kullback-Leibler divergence 是非负的,并且只有当两个分布完全相同的时候才会取到 0 。因此我们可以得到关系?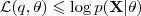 ?,亦即?
?,亦即?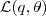 ?是?
?是?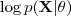 ?的一个下界。
?的一个下界。
现在考虑 EM 的迭代过程,记上一次迭代得出的参数为?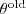 ,现在我们要选取?
,现在我们要选取?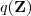 ?以令?
?以令?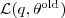 ?最大,由于?
?最大,由于?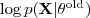 ?并不依赖于?
?并不依赖于?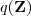 ?,因此?
?,因此?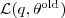 ?的上限(在?
?的上限(在?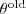 ?固定的时候)是一个定值,它取到这个最大值的条件就是 Kullback-Leibler divergence 为零,亦即?
?固定的时候)是一个定值,它取到这个最大值的条件就是 Kullback-Leibler divergence 为零,亦即?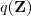 ?等于后验概率?
?等于后验概率?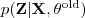 ?。把它带入到?
?。把它带入到?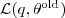 ?的表达式中可以得到
?的表达式中可以得到
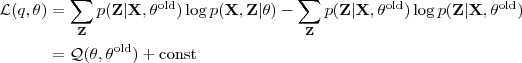
其中?const?是常量,而?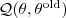 ?则正是我们之前所得到的"同时包含了 sample 和隐含变量的 Log-likelihood function 关于后验概率的期望",因此这个对应到 EM 中的"E"一步。
?则正是我们之前所得到的"同时包含了 sample 和隐含变量的 Log-likelihood function 关于后验概率的期望",因此这个对应到 EM 中的"E"一步。
在接下来的"M"一步中,我们讲固定住分布?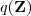 ?,再选取合适的?
?,再选取合适的? ?以将?
?以将?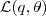 ?最大化,这次其上界?
?最大化,这次其上界?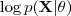 ?也依赖于变量?
?也依赖于变量? ?,并会随着?
?,并会随着?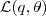 ?的增大而增大(因为我们有前面的不等式成立)。一般情况下?
?的增大而增大(因为我们有前面的不等式成立)。一般情况下?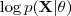 ?增大的量会比?
?增大的量会比?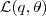 ?要多一些,这时候 Kullback-Leibler divergence 在新的参数?
?要多一些,这时候 Kullback-Leibler divergence 在新的参数? ?下又不为零了,因此我们可以进入下一轮迭代,重新回到"E"一步去求新的?
?下又不为零了,因此我们可以进入下一轮迭代,重新回到"E"一步去求新的?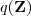 ?;另一方面,如果这里 Kullback-Leibler divergence 在新的参数下还是等于 0 ,那么说明我们已经达到了一个(至少是局部的)最优解,迭代过程可以结束了。
?;另一方面,如果这里 Kullback-Leibler divergence 在新的参数下还是等于 0 ,那么说明我们已经达到了一个(至少是局部的)最优解,迭代过程可以结束了。
上面的推导中我们可以看到每一次迭代 E 和 M 两个步骤都是在对解进行改进,因此迭代的过程中得到的 likelihood 会逐渐逼近(至少是局部的)最优值。另外,在 M 一步中除了用最大似然之外,也可以引入先验使用?Maximum a Posteriori (MAP)?的方法来做。还有一些很困难的问题,甚至在迭代的过程中 M 一步也不能直接求出最大值,这里我们通过把要求放宽──并不一定要求出最大值,只要能够得到比旧的值更好的结果即可,这样的做法通常称作 Generalized EM (GEM)。
当然,一开始我们就说了,EM 是一个通用的算法,并不只是用来求解 GMM 。下面我们就来举一个用 EM 来做中文分词的例子。这个例子来源于论文?Self-supervised Chinese Word Segmentation?。我在上次 MSTC 搜索引擎系列小课堂讲文本处理的时候提到过。这里为了把注意力集中到 EM 上,我略去一些细节的东西,简单地描述一下基本的模型。
现在我们有一个字符序列?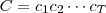 ,并希望得到一个模型?
,并希望得到一个模型?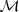 (依赖于参数?
(依赖于参数? )能用来将其词的序列?
)能用来将其词的序列?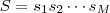 ?。按照生成模型的方式来考虑,将?
?。按照生成模型的方式来考虑,将? ?看成是由?
?看成是由?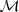 ?生成的序列的话,我们自然希望找到那个生成?
?生成的序列的话,我们自然希望找到那个生成? ?的可能性最大的?
?的可能性最大的?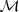 ?,亦即通过最大似然的方式来估计参数?
?,亦即通过最大似然的方式来估计参数? ?。
?。
然而我们不知道似然函数?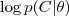 ?应该如何去优化,因此我们引入 latent variable?
?应该如何去优化,因此我们引入 latent variable?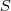 ?,如果我们知道?
?,如果我们知道?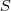 ?的话,似然值很容易求得:
?的话,似然值很容易求得:
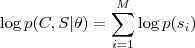
其中?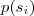 ?的值是从模型?
?的值是从模型?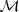 ?中已知的。但是现在我们不知道?
?中已知的。但是现在我们不知道?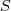 ?的值,因此我们转而取其关于后验概率的期望:
?的值,因此我们转而取其关于后验概率的期望:
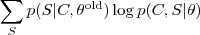
然后将这个期望针对? ?最大化即完成了 EM 的一次迭代。具体的做法通常是先把一个初始文本(比如所有的?
?最大化即完成了 EM 的一次迭代。具体的做法通常是先把一个初始文本(比如所有的? ?的集合)按照 N-gram 分割(N-gram 在?讲 K-medoids 的那篇文章中介绍过)为?
?的集合)按照 N-gram 分割(N-gram 在?讲 K-medoids 的那篇文章中介绍过)为?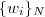 ,形成一个最初的辞典,而模型?
,形成一个最初的辞典,而模型?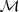 ?的参数?
?的参数? ?实际上就描述了各个 N-gram 的概率?
?实际上就描述了各个 N-gram 的概率?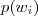 ?,初始值可以直接取为频率值。有了辞典之后对于任意的?
?,初始值可以直接取为频率值。有了辞典之后对于任意的? ?,我们可以根据辞典枚举出所有可能的分割?
?,我们可以根据辞典枚举出所有可能的分割?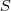 ?,而每个分割的后验概率?
?,而每个分割的后验概率?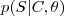 ?就是其中的单词的概率乘积。其他的就是标准的 EM 算法的迭代步骤了。
?就是其中的单词的概率乘积。其他的就是标准的 EM 算法的迭代步骤了。
实际上在实际产品中我们会使用一些带了更多启发式规则的分词方法(比如?MMSEG),而这个方法更大的用处在于从一堆文本中"学习"出一个词典来(也就是?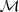 ?),并且这个过程是全自动的,主要有两个优点:
?),并且这个过程是全自动的,主要有两个优点:
不管怎样,以上两点看起来都非常诱人。不过理论离实际往往还是有不少差距的。我不知道实际分词系统中有没有在用这样的方法来做训练的。之前我曾经用 Wikipedia (繁体中文)上抓下来的一些数据跑过一下小规模的试验,结果还可以,但是也并不如想像中的完美。因为当时也并没有弄明白 EM 是怎么一回事,加上这个过程本身计算负荷还是非常大的,也就没有深入下去。也许以后有时间可以再尝试一下。
总之,EM 是一种应用广泛的算法,虽然讲了点理论也讲了例子,但是一没有贴代码二没有贴图片,似乎实在是不像我的作风,不过精力和篇幅有限,也只能到此为止了。
如果说?K-means?和?GMM?这些聚类的方法是古代流行的算法的话,那么这次要讲的 Spectral Clustering 就可以算是现代流行的算法了,中文通常称为"谱聚类"。由于使用的矩阵的细微差别,谱聚类实际上可以说是一"类"算法。
Spectral Clustering 和传统的聚类方法(例如 K-means)比起来有不少优点:
突然冒出这么一个要求比 K-means 要少,结果比 K-means 要好,算得还比 K-means 快的东西,实在是让人不得不怀疑是不是江湖骗子啊。所以,是骡子是马,先拉出来溜溜再说。不过,在K-medoids?那篇文章中曾经实际跑过 K-medoids 算法,最后的结果也就是一个 accuracy ,一个数字又不能画成图表之类的,看起来实在是没意思,而且 K-means 跑起来实在是太慢了,所以这里我还是稍微偷懒一下,直接引用一下一篇论文里的结果吧。
结果来自论文?Document clustering using locality preserving indexing?这篇论文,这篇论文实际上是提的另一种聚类方法(下次如果有机会也会讲到),不过在它的实验中也有 K-means 和 Spectral Clustering 这两组数据,抽取出来如下所示:
k | TDT2 | Reuters-21578 | ||
K-means | SC | K-means | SC | |
2 | 0.989 | 0.998 | 0.871 | 0.923 |
3 | 0.974 | 0.996 | 0.775 | 0.816 |
4 | 0.959 | 0.996 | 0.732 | 0.793 |
… | ||||
9 | 0.852 | 0.984 | 0.553 | 0.625 |
10 | 0.835 | 0.979 | 0.545 | 0.615 |
其中 TDT2 和 Reuters-21578 分别是两个被广泛使用的标准文本数据集,虽然在不同的数据集上得出来的结果并不能直接拿来比较,但是在同一数据集上 K-means 和 SC (Spectral Clustering) 的结果对比就一目了然了。实验中分别抽取了这两个数据集中若干个类别(从 2 类到 10 类)的数据进行聚类,得出的 accuracy 分别在上表中列出(我偷懒没有全部列出来)。可以看到,Spectral Clustering 这里完胜 K-means 。

这么强大的算法,再戴上"谱聚类"这么一个高深莫测的名号,若不是模型无比复杂、包罗宇宙,就肯定是某镇山之宝、不传秘籍吧?其实不是这样,Spectral Clustering 不管从模型上还是从实现上都并不复杂,只需要能求矩阵的特征值和特征向量即可──而这是一个非常基本的运算,任何一个号称提供线性代数运算支持的库都理应有这样的功能。而关于 Spectral Clustering 的秘籍更是满街都是,随便从地摊上找来一本,翻开便可以看到 Spectral Clustering 算法的全貌:
 ?。一个最偷懒的办法就是:直接用我们前面在 K-medoids 中用的相似度矩阵作为?
?。一个最偷懒的办法就是:直接用我们前面在 K-medoids 中用的相似度矩阵作为? ?。
?。
 ?的每一列元素加起来得到?
?的每一列元素加起来得到?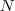 ?个数,把它们放在对角线上(其他地方都是零),组成一个?
?个数,把它们放在对角线上(其他地方都是零),组成一个?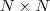 ?的矩阵,记为?
?的矩阵,记为?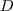 ?。并令?
?。并令?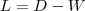 ?。
?。
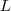 ?的前?
?的前?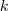 ?个特征值(在本文中,除非特殊说明,否则"前?
?个特征值(在本文中,除非特殊说明,否则"前?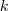 ?个"指按照特征值的大小从小到大的顺序)
?个"指按照特征值的大小从小到大的顺序)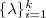 ?以及对应的特征向量?
?以及对应的特征向量?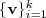 ?。
?。
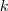 ?个特征(列)向量排列在一起组成一个?
?个特征(列)向量排列在一起组成一个?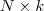 ?的矩阵,将其中每一行看作?
?的矩阵,将其中每一行看作?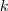 ?维空间中的一个向量,并使用 K-means 算法进行聚类。聚类的结果中每一行所属的类别就是原来 Graph 中的节点亦即最初的?
?维空间中的一个向量,并使用 K-means 算法进行聚类。聚类的结果中每一行所属的类别就是原来 Graph 中的节点亦即最初的?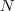 ?个数据点分别所属的类别。
?个数据点分别所属的类别。
就是这么几步,把数据做了一些诡异的变换,然后还在背后偷偷地调用了 K-means 。到此为止,你已经可以拿着它上街去招摇撞骗了。不过,如果你还是觉得不太靠谱的话,不妨再接着往下看,我们再来聊一聊 Spectral Clustering 那几个"诡异变换"背后的道理何在。
其实,如果你熟悉?Dimensional Reduction?(降维)的话,大概已经看出来了,Spectral Clustering 其实就是通过 Laplacian Eigenmap 的降维方式降维之后再做 K-means 的一个过程──听起来土多了。不过,为什么要刚好降到?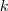 ?维呢?其实整个模型还可以从另一个角度导出来,所以,让我们不妨先跑一下题。
?维呢?其实整个模型还可以从另一个角度导出来,所以,让我们不妨先跑一下题。
在 Image Processing (我好像之前有听说我对这个领域深恶痛绝?)里有一个问题就是对图像进行?Segmentation?(区域分割),也就是让相似的像素组成一个区域,比如,我们一般希望一张照片里面的人(前景)和背景被分割到不同的区域中。在 Image Processing 领域里已经有许多自动或半自动的算法来解决这个问题,并且有不少方法和 Clustering 有密切联系。比如我们在谈?Vector Quantization?的时候就曾经用 K-means 来把颜色相似的像素聚类到一起,不过那还不是真正的 Segmentation ,因为如果仅仅是考虑颜色相似的话,图片上位置离得很远的像素也有可能被聚到同一类中,我们通常并不会把这样一些"游离"的像素构成的东西称为一个"区域",但这个问题其实也很好解决:只要在聚类用的 feature 中加入位置信息(例如,原来是使用 R、G、B 三个值来表示一个像素,现在加入 x、y 两个新的值)即可。
另一方面,还有一个经常被研究的问题就是?Graph Cut?,简单地说就是把一个 Graph 的一些边切断,让他被打散成一些独立联通的 sub-Graph ,而这些被切断的边的权值的总和就被称为?Cut值。如果用一张图片中的所有像素来组成一个 Graph ,并把(比如,颜色和位置上)相似的节点连接起来,边上的权值表示相似程度,那么把图片分割为几个区域的问题实际上等价于把 Graph 分割为几个 sub-Graph 的问题,并且我们可以要求分割所得的 Cut 值最小,亦即:那些被切断的边的权值之和最小,直观上我们可以知道,权重比较大的边没有被切断,表示比较相似的点被保留在了同一个 sub-Graph 中,而彼此之间联系不大的点则被分割开来。我们可以认为这样一种分割方式是比较好的。
实际上,抛开图像分割的问题不谈,在 Graph Cut 相关的一系列问题中,Minimum cut (最小割)本身就是一个被广泛研究的问题,并且有成熟的算法来求解。只是单纯的最小割在这里通常并不是特别适用,很多时候只是简单地把和其他像素联系最弱的那一个像素给分割出去了,相反,我们通常更希望分割出来的区域(的大小)要相对均匀一些,而不是一些很大的区块和一些几乎是孤立的点。为此,又有许多替代的算法提出来,如 Ratio Cut 、Normalized Cut 等。
不过,在继续讨论之前,我们还是先来定义一下符号,因为仅凭文字还是很难表述清楚。将 Graph 表示为邻接矩阵的形式,记为? ,其中?
,其中?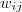 ?是节点?
?是节点? ?到节点?
?到节点?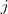 ?的权值,如果两个节点不是相连的,权值为零。设?
?的权值,如果两个节点不是相连的,权值为零。设?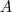 ?和?
?和?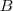 ?为 Graph 的两个子集(没有交集),那么两者之间的 cut 可以正式定义为:
?为 Graph 的两个子集(没有交集),那么两者之间的 cut 可以正式定义为:
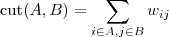
先考虑最简单的情况,如果把一个 Graph 分割为两个部分的话,那么 Minimum cut 就是要最小化?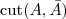 ?(其中?
?(其中?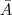 ?表示?
?表示?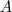 ?的补集)。但是由于这样经常会出现孤立节点被分割出来的情况,因此又出现了 RatioCut :
?的补集)。但是由于这样经常会出现孤立节点被分割出来的情况,因此又出现了 RatioCut :
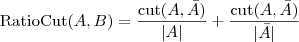
以及 NormalizedCut :
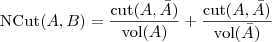
其中?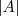 ?表示?
?表示?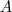 ?中的节点数目,而?
?中的节点数目,而?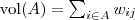 ?。两者都可以算作?
?。两者都可以算作?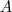 ?的"大小"的一种度量,通过在分母上放置这样的项,就可以有效地防止孤立点的情况出现,达到相对平均一些的分割。事实上,Jianbo Shi?的这篇 PAMI paper:Normalized Cuts and Image Segmentation?正是把 NormalizedCut 用在图像分割上了。
?的"大小"的一种度量,通过在分母上放置这样的项,就可以有效地防止孤立点的情况出现,达到相对平均一些的分割。事实上,Jianbo Shi?的这篇 PAMI paper:Normalized Cuts and Image Segmentation?正是把 NormalizedCut 用在图像分割上了。
搬出 RatioCut 和 NormalizedCut 是因为它们和这里的 Spectral Clustering 实际上有非常紧密的联系。看看 RatioCut ,式子虽然简单,但是要最小化它却是一个 NP 难问题,不方便求解,为了找到解决办法,让我们先来做做变形。
令?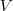 ?表示 Graph 的所有节点的集合,首先定义一个?
?表示 Graph 的所有节点的集合,首先定义一个?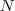 ?维向量?
?维向量?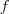 :
:
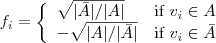
再回忆一下我们最开始定义的矩阵?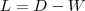 ?,其实它有一个名字,叫做 Graph Laplacian ,不过,我们后面可以看到,其实有好几个类似的矩阵都叫做这个名字:
?,其实它有一个名字,叫做 Graph Laplacian ,不过,我们后面可以看到,其实有好几个类似的矩阵都叫做这个名字:
Usually, every author just calls "his" matrix the graph Laplacian.
其实也可以理解,就好象现在所有的厂家都说自己的技术是"云计算"一样。这个?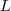 ?有一个性质就是:
?有一个性质就是:
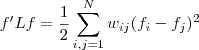
这个是对任意向量?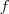 ?都成立的,很好证明,只要按照定义展开就可以得到了。把我们刚才定义的那个?
?都成立的,很好证明,只要按照定义展开就可以得到了。把我们刚才定义的那个?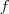 ?带进去,就可以得到
?带进去,就可以得到
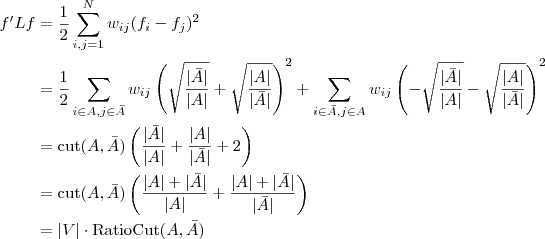
另外,如果令?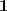 ?为各个元素全为 1 的向量的话,直接展开可以很容易得到?
?为各个元素全为 1 的向量的话,直接展开可以很容易得到?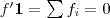 ?和?
?和?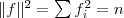 ?。由于?
?。由于?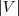 ?是一个常量,因此最小化 RatioCut 就等价于最小化?
?是一个常量,因此最小化 RatioCut 就等价于最小化?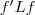 ?,当然,要记得加上附加条件?
?,当然,要记得加上附加条件?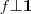 ?以及?
?以及?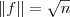 ?。
?。
问题转化到这个样子就好求了,因为有一个叫做?Rayleigh quotient?的东西:
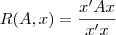
他的最大值和最小值分别等于矩阵?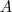 ?的最大的那个特征值和最小的那个特征值,并且极值在?
?的最大的那个特征值和最小的那个特征值,并且极值在? 等于对应的特征向量时取到。由于?
等于对应的特征向量时取到。由于?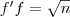 ?是常数,因此最小化?
?是常数,因此最小化?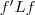 ?实际上也就等价于最小化?
?实际上也就等价于最小化?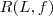 ?,不过由于?
?,不过由于?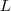 ?的最小的特征值为零,并且对应的特征向量正好为?
?的最小的特征值为零,并且对应的特征向量正好为?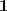 ?(我们这里仅考虑 Graph 是联通的情况),不满足?
?(我们这里仅考虑 Graph 是联通的情况),不满足?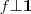 ?的条件,因此我们取第二个小的特征值,以及对应的特征向量?
?的条件,因此我们取第二个小的特征值,以及对应的特征向量? ?。
?。
到这一步,我们看起来好像是很容易地解决了前面那个 NP 难问题,实际上是我们耍了一个把戏:之前的问题之所以 NP 难是因为向量?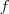 ?的元素只能取两个值?
?的元素只能取两个值?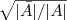 ?和?
?和? ?中的一个,是一个离散的问题,而我们求的的特征向量?
?中的一个,是一个离散的问题,而我们求的的特征向量? ?其中的元素可以是任意实数,就是说我们将原来的问题限制放宽了。那如何得到原来的解呢?一个最简单的办法就是看?
?其中的元素可以是任意实数,就是说我们将原来的问题限制放宽了。那如何得到原来的解呢?一个最简单的办法就是看? ?的每个元素是大于零还是小于零,将他们分别对应到离散情况的?
?的每个元素是大于零还是小于零,将他们分别对应到离散情况的?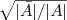 ?和?
?和? ?,不过我们也可以采取稍微复杂一点的办法,用?
?,不过我们也可以采取稍微复杂一点的办法,用?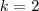 ?的 K-means 来将?
?的 K-means 来将? ?的元素聚为两类。
?的元素聚为两类。
到此为止,已经有 Spectral Clustering 的影子了:求特征值,再对特征向量进行 K-means 聚类。实际上,从两类的问题推广到 k 类的问题(数学推导我就不再详细写了),我们就得到了和之前的 Spectral Clustering 一模一样的步骤:求特征值并取前 k 个最小的,将对应的特征向量排列起来,再按行进行 K-means 聚类。分毫不差!
用类似的办法,NormalizedCut 也可以等价到 Spectral Clustering 不过这次我就不再讲那么多了,感兴趣的话(还包括其他一些形式的 Graph Laplacian 以及 Spectral Clustering 和?Random walk?的关系),可以去看这篇 Tutorial :A Tutorial on Spectral Clustering?。
为了缓和一下气氛,我决定贴一下 Spectral Clustering 的一个简单的 Matlab 实现:
? ? ? ? |
最后,我们再来看一下本文一开始说的 Spectral Clustering 的几个优点:
 ?中。不过一般?
?中。不过一般? ?并不总是等于最初的相似度矩阵——回忆一下,
?并不总是等于最初的相似度矩阵——回忆一下, 是我们构造出来的 Graph 的邻接矩阵表示,通常我们在构造 Graph 的时候为了方便进行聚类,更加强到"局部"的连通性,亦即主要考虑把相似的点连接在一起,比如,我们设置一个阈值,如果两个点的相似度小于这个阈值,就把他们看作是不连接的。另一种构造 Graph 的方法是将 n 个与节点最相似的点与其连接起来。
是我们构造出来的 Graph 的邻接矩阵表示,通常我们在构造 Graph 的时候为了方便进行聚类,更加强到"局部"的连通性,亦即主要考虑把相似的点连接在一起,比如,我们设置一个阈值,如果两个点的相似度小于这个阈值,就把他们看作是不连接的。另一种构造 Graph 的方法是将 n 个与节点最相似的点与其连接起来。
说了这么多,好像有些乱,不过也只能到此打住了。最后再多嘴一句,Spectral Clustering 名字来源于?Spectral theory?,也就是用特征分解来分析问题的理论了。
UPDATE 2011.11.23: 有不少同学问我关于代码的问题,这里更新两点主要的问题:
由于总是有各种各样的杂事,这个系列的文章竟然一下子拖了好几个月,(实际上其他的日志我也写得比较少),现在决定还是先把这篇降维的日志写完。我甚至都以及忘记了在这个系列中之前有没有讲过"特征"(feature)的概念了,这里不妨再稍微提一下。机器学习应用到各个领域里,会遇到许多不同类型的数据要处理:图像、文本、音频视频以及物理、生物、化学等实验还有其他工业、商业以及军事上得到的各种数据,如果要为每一种类型的数据都设计独立的算法,那显然是非常不现实的事,因此,机器学习算法通常会采用一些标准的数据格式,最常见的一种格式就是每一个数据对应欧几里德空间里的一个向量。
如果原始的数据格式不兼容,那么就需要首先进行转换,这个过程通常叫做"特征提取"(Feature Extraction),而得到的标准数据格式通常叫做 Feature 。例如,一个最简单的将一个文本 Document 转化为向量的方法如下:
当然,在实际中我们几乎不会这样人工设定空间的各个维度所对应的单词,而通常是从一个数据集中统计出所有出现的词,再将其中的一些挑选出来作为维度。怎样挑选呢?最简单的办法是根本不做任何挑选,或者简单地只是把出现频率太低的单词(维度)去掉。
不过,事实上我们通常会做更复杂一些的处理,例如,如果你是在做?sentiment analysis?,那么你通常会更加关注语气很重的词,比如 "bad"、"terrible"、"awesome" 等的重要性就比普通的词要大,此时你可以为每一个维度设一个权重,例如,给 "bad" 设置权重 2 ,那么出现 3 次的话,向量在该维度对应的值就为?2*3 = 6?。当然这样手工指定权重只在小范围内可行,如果要为数百万个维度指定权重,那显然是不可能的,另一个稍微自动一点的办法是?tf-idf?。
tf 就是 Term Frequency ,就和刚才说的单词出现的次数差不多,而 idf 则是 Inverse Document Frequency ,通常使用如下公式进行计算:
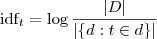
这相当于自动计算出来的每个单词的权重,其想法很简单:如果在许多文档中都出现的词(例如虚词、语气词等),它所包含的信息量通常会比较小,所以用以上的公式计算出来的权重也会相对较小;另一方面,如果单词并不是在很多文档中都出现的话,很有可能就是出现的那些文档的重要特征,因此权重会相对大一些。
前面说了一堆,其实主要是想要对 feature 做一些"预"处理,让它更"好"一些,手工设置维度的权重固然是很人力,其实 tf-idf 也是在假定了原始 feature 是 document 、term 这样的形式(或者类似的模型)的情况下才适用的(例如,在门禁之类的系统里,feature 可能有声音、人脸图像以及指纹等数据,就不能这样来处理),因此也并不能说是一种通用的方法。
然而,既然机器学习的算法可以在不考虑原始数据的来源和意义的情况下工作,那么 feature 的处理应该也可以。事实也是如此,包括 feature selection 和 dimensionality reduction 两个 topic 都有许多很经典的算法。前者通常是选出重要的 feature 的维度(并抛弃不重要的维度),而后者则是更广泛意义上的把一个高维的向量映射为一个低维向量,亦即"降维",得到的结果 feature 的值已经不一定是原始的值,也可以把 feature selection 看作是 dimensionality reduction 的一种特殊情况。举一个例子,tf-idf 实际上不算 feature selection ,因为它(通常)并没有丢弃低权值的维度,并且处理过后的特征的每个维度都被乘上了一个权值,不再是原来的值了;但是它却可以被看作一种降维,虽然严格意义上来说维度并没有"降低"。简单来说降维可以看作一个函数,其输入是一个 D 维的向量,输出是一个 M 维的向量。
按照机器学习的方法的一贯作风,我们需要定义目标函数并进行最优化。不同的目标也就导致了不同的降维算法,这也正是今天要讲的话题。
然而,我们的目的究竟是什么呢?一个比较直接的问题是原始的数据量维度太高,我们无法处理,因此需要降维,此时我们通常希望在最大限度地降低数据的维度的前提下能够同时保证保留目标的重要的信息,就好比在做有损的数据压缩时希望压缩比尽量大但是质量损失不要太多。于是问题又转化为如何衡量对信息的保留程度了。
一个最直接的办法就是衡量 reconstruction error ,即
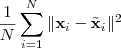
其中?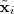 ?是??所对应的低维表示再重新构造出来的高维形式,就相当于是压缩之后解压出来的结果,不过虽然有许多压缩方法都是无损的,就是说这个差值会等于零,但是大部分降维的结果都是有损的。不过我们仍然希望把上面的 reconstruction error 最小化。
?是??所对应的低维表示再重新构造出来的高维形式,就相当于是压缩之后解压出来的结果,不过虽然有许多压缩方法都是无损的,就是说这个差值会等于零,但是大部分降维的结果都是有损的。不过我们仍然希望把上面的 reconstruction error 最小化。
另外一种方式是简单地使用?variance?来衡量所包含信息量,例如,我们要把一些 D 维的向量降为 1 维,那么我们希望这一维的 variance 达到最大化,亦即:
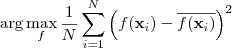
其中?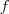 ?是降维函数。推而广之,如果是降为 2 维,那么我希望第 2 维去关注第 1 维之外的 variance ,所以要求它在与第一维垂直的情况下也达到 variance 最大化。以此类推。
?是降维函数。推而广之,如果是降为 2 维,那么我希望第 2 维去关注第 1 维之外的 variance ,所以要求它在与第一维垂直的情况下也达到 variance 最大化。以此类推。
然而,当我们把降维函数?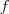 ?限定维线性的时候,两种途径会得到同样的结果,就是被广泛使用的?Principal Components Analysis(PCA) 。PCA 的降维函数是线性的,可以用一个?
?限定维线性的时候,两种途径会得到同样的结果,就是被广泛使用的?Principal Components Analysis(PCA) 。PCA 的降维函数是线性的,可以用一个?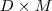 ?维的矩阵?
?维的矩阵? ?来表示,因此,一个 D 维的向量?
?来表示,因此,一个 D 维的向量? ?经过线性变换?
?经过线性变换?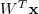 ?之后得到一个 M 维向量,就是降维的结果。把原始数据按行排列为一个?
?之后得到一个 M 维向量,就是降维的结果。把原始数据按行排列为一个?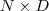 ?维的矩阵?
?维的矩阵?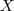 ?,则?
?,则?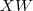 ?就是降维后的?
?就是降维后的?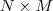 ?维的数据矩阵,目标是使其?covariance?矩阵最大。在数据被规则化(即减去其平均值)过的情况下,协方差矩阵 (covariance)?
?维的数据矩阵,目标是使其?covariance?矩阵最大。在数据被规则化(即减去其平均值)过的情况下,协方差矩阵 (covariance)?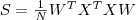 ?,当然矩阵不是一个数,不能直接最大化,如果我们采用矩阵的?Trace?(亦即其对角线上元素的和)来衡量其大小的话,要对?
?,当然矩阵不是一个数,不能直接最大化,如果我们采用矩阵的?Trace?(亦即其对角线上元素的和)来衡量其大小的话,要对?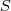 ?求最大化,只需要求出?
?求最大化,只需要求出?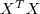 ?的特征值和特征向量,将 M 个最大的特征值所对应的特征向量按列排列起来组成线性变换矩阵?
?的特征值和特征向量,将 M 个最大的特征值所对应的特征向量按列排列起来组成线性变换矩阵? ?即可。这也就是 PCA 的求解过程,得到的降维矩阵?
?即可。这也就是 PCA 的求解过程,得到的降维矩阵? 可以直接用到新的数据上。如果熟悉?Latent Semantic Analysis?(LSA) 的话,大概已经看出 PCA 和?Singular Value Decomposition?(SVD) 以及 LSA 之间的关系了。以下这张图(引自《The Elements of Statistical Learning》)可以直观地看出来 PCA 做了什么,对于一些原始为二维的数据,PCA 首先找到了 variance 最大的那一个方向:
可以直接用到新的数据上。如果熟悉?Latent Semantic Analysis?(LSA) 的话,大概已经看出 PCA 和?Singular Value Decomposition?(SVD) 以及 LSA 之间的关系了。以下这张图(引自《The Elements of Statistical Learning》)可以直观地看出来 PCA 做了什么,对于一些原始为二维的数据,PCA 首先找到了 variance 最大的那一个方向:
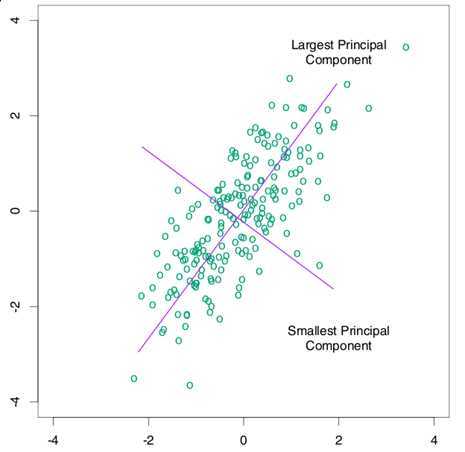
PCA 作为一种经典的降维方法,被广泛地应用于机器学习、计算机视觉以及信息检索等各个领域,其地位类似于聚类中的 K-means ,在现在关于降维等算法的研究中也经常被作为 baseline 出现。然而,PCA 也有一些比较明显的缺点:一个就是 PCA 降维是线性变换,虽然线性变换计算方便,并且可以很容易地推广到新的数据上,然而有些时候线性变换也许并不合适,为此有许多扩展被提出来,其中一个就是?Kernel PCA?,用?Kernel Trick?来将 PCA 推广到非线性的情况。另外,PCA 实际上可以看作是一个具有 Gaussian 先验和条件概率分布的 latent variable 模型,它假定数据的 mean 和 variance 是重要的特征,并依靠 covariance 最大化来作为优化目标,而事实上这有时候对于解决问题帮助并不大。
一个典型的问题就是做聚类或分类,回想我们之前谈过的?Spectral Clustering?,就是使用 Laplacian eigenmap 降维之后再做 K-means 聚类,如果问题定下来了要对数据进行区分的话,"目的"就变得明朗了一些,也就是为了能够区分不同类别的数据,再考虑直观的情况,我们希望如果通过降维把高维数据变换到一个二维平面上的话,可以很容易"看"出来不同类别的数据被映射到了不同的地方。虽然 PCA 极力降低 reconstruction error ,试图得到可以代表原始数据的 components ,但是却无法保证这些 components 是有助于区分不同类别的。如果我们有训练数据的类别标签,则可以用?Fisher Linear Discriminant Analysis?来处理这个问题。
同 PCA 一样,Fisher Linear Discriminant Analysis 也是一个线性映射模型,只不过它的目标函数并不是 Variance 最大化,而是有针对性地使投影之后属于同一个类别的数据之间的 variance 最小化,并且同时属于不同类别的数据之间的 variance 最大化。具体的形式和推导可以参见《Pattern Classification》这本书的第三章?Component Analysis and Discriminants?。
当然,很多时候(比如做聚类)我们并不知道原始数据是属于哪个类别的,此时 Linear Discriminant Analysis 就没有办法了。不过,如果我们假设原始的数据形式就是可区分的的话,则可以通过保持这种可区分度的方式来做降维,MDS?是 PCA 之外的另一种经典的降维方法,它降维的限制就是要保持数据之间的相对距离。实际上 MDS 甚至不要求原始数据是处在一个何种空间中的,只要给出他们之间的相对"距离",它就可以将其映射到一个低维欧氏空间中,通常是三维或者二维,用于做 visualization 。
不过我在这里并不打算细讲 MDS ,而是想说一下在 Spectral Clustering 中用到的降维方法 Laplacian Eigenmap 。同 MDS 类似,LE 也只需要有原始数据的相似度矩阵,不过这里通常要求这个相似度矩阵?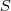 ?具有局部性质,亦即只考虑局部领域内的相似性,如果点?
?具有局部性质,亦即只考虑局部领域内的相似性,如果点? ?和?
?和?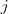 ?距离太远的话,
?距离太远的话,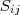 ?应该为零。有两种很直接的办法可以让普通的相似度矩阵具有这种局部性质:
?应该为零。有两种很直接的办法可以让普通的相似度矩阵具有这种局部性质:
 -领域内考虑局部性。
-领域内考虑局部性。
 ?属于?
?属于?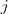 ?的 k 个最接近的邻居且/或反之的时候,就保留?
?的 k 个最接近的邻居且/或反之的时候,就保留?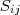 ?的值,否则置为零。
?的值,否则置为零。
构造好?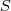 ?之后再来考虑降维,从最简单的情况开始,即降到一维?
?之后再来考虑降维,从最简单的情况开始,即降到一维?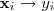 ?,通过最小化如下目标函数来实现:
?,通过最小化如下目标函数来实现:
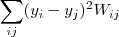
从直观上来说,这样的目标函数的意义在于:如果原来?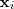 ?和?
?和?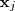 ?比较接近,那么?
?比较接近,那么?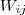 ?会相对比较大,这样如果映射过后?
?会相对比较大,这样如果映射过后?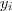 ?和?
?和?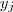 ?相差比较大的话,就会被权重?
?相差比较大的话,就会被权重?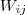 ?放大,因此最小化目标函数就保证了原来相近的点在映射过后也不会彼此相差太远。
?放大,因此最小化目标函数就保证了原来相近的点在映射过后也不会彼此相差太远。
令?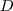 ?为将?
?为将? ?的每一行加起来所得到的对角阵,而?
?的每一行加起来所得到的对角阵,而?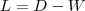 ?,被称作是拉普拉斯矩阵,通过求解如下的特征值问题
?,被称作是拉普拉斯矩阵,通过求解如下的特征值问题
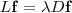
易知最小的那个特征值肯定是 0 ,除此之外的最小的特征值所对应的特征向量就是映射后的结果。特征向量是一个 N 维列向量,将其旋转一下,正好是 N 个原始数据降到一维之后的结果。
类似地推广到 M 维的情况,只需要取除去 0 之外的最小的 M 个特征值所对应的特征向量,转置之后按行排列起来,就是降维后的结果。用 Matlab 代码写出来如下所示(使用了 knn 来构造相似度矩阵,并且没有用 heat kernel ):
| ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? |
事实上,Laplacian Eigenmap 假设数据分布在一个嵌套在高维空间中的低维流形上, Laplacian Matrix? ?则是流形的?Laplace Beltrami operator?的一个离散近似。关于流形以及 Laplacian Eigenmap 这个模型的理论知识就不在这里做更多地展开了,下面看一个比较直观的例子 Swiss Roll 。
?则是流形的?Laplace Beltrami operator?的一个离散近似。关于流形以及 Laplacian Eigenmap 这个模型的理论知识就不在这里做更多地展开了,下面看一个比较直观的例子 Swiss Roll 。
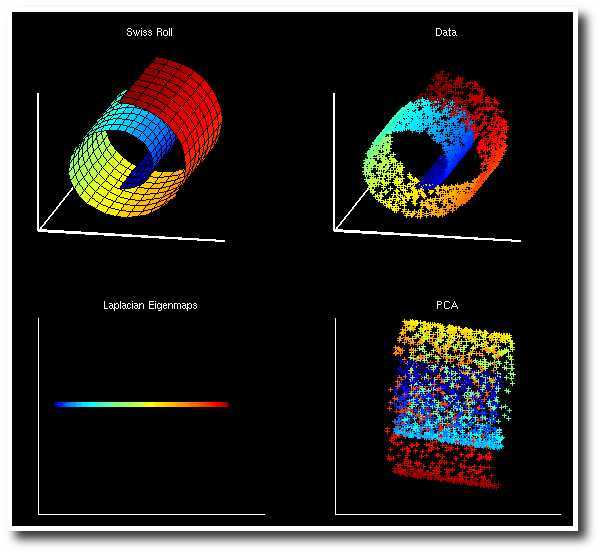
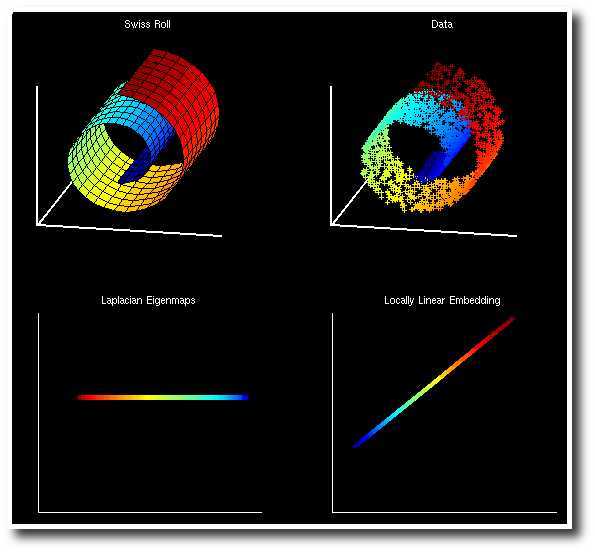
Swiss Roll 是一个像面包圈一样的结构,可以看作一个嵌套在三维空间中的二维流形,如下图所示,左边是 Swiss Roll ,右边是从 Swiss Roll 中随机选出来的一些点,为了标明点在流形上的位置,给它们都标上了颜色。
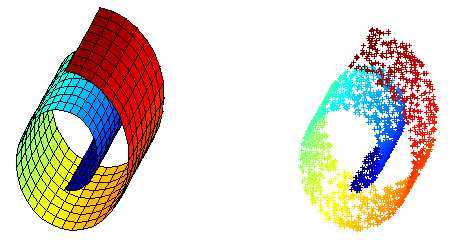
而下图是 Laplacian Eigenmap 和 PCA 分别对 Swiss Roll 降维的结果,可以看到 LE 成功地把这个流形展开把在流形上属于不同位置的点映射到了不同的地方,而 PCA 的结果则很糟糕,几种颜色都混杂在了一起。
另外,还有一种叫做 Locally Linear Embedding 的降维方法,它同 LE 一样采用了流形假设,并假定平滑流形在局部具有线性性质,一个点可以通过其局部邻域内的点重构出来。首先它会将下式最小化
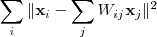
以求解出最优的局部线性重构矩阵? ?,对于距离较远的点?
?,对于距离较远的点? ?和?
?和?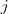 ?,
?,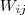 ?应当等于零。这之后再把?
?应当等于零。这之后再把? ?当作已知量对下式进行最小化:
?当作已知量对下式进行最小化:
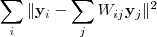
这里?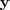 ?成了变量,亦即降维后的向量,对这个式子求最小化的意义很明显了,就是要求如果原来的数据?
?成了变量,亦即降维后的向量,对这个式子求最小化的意义很明显了,就是要求如果原来的数据?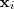 ?可以以?
?可以以? ?矩阵里对应的系数根据其邻域内的点重构出来的话,那么降维过后的数据也应该保持这个性质。
?矩阵里对应的系数根据其邻域内的点重构出来的话,那么降维过后的数据也应该保持这个性质。
经过一些变换之后得到的求解方法和 LE 类似,也是要求解一个特征值问题,实际上,从理论上也可以得出二者之间的联系(LE 对应于?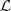 ?而 LLE 对应于?
?而 LLE 对应于?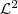 ),如果感兴趣的话,可以参考Laplacian Eigenmaps for Dimensionality Reduction and Data Representation?这篇论文里的对比。下面是两种方法在 Swiss Roll 数据上的结果,也是非常相似的:
),如果感兴趣的话,可以参考Laplacian Eigenmaps for Dimensionality Reduction and Data Representation?这篇论文里的对比。下面是两种方法在 Swiss Roll 数据上的结果,也是非常相似的:
有一点需要注意的是,LE 和 LLE 都是非线性的方法,PCA 得到的结果是一个投影矩阵,这个结果可以保存下来,在之后对任意向量进行投影,而 LE 和 LLE都是直接得出了数据降维之后的结果,但是对于新的数据,却没有得到一个"降维函数",没有一个合适的方法得到新的数据的降维结果。所以,在人们努力寻求非线性形式扩展 PCA 的时候,另一些人则提出了 LE 和 LLE 的线性形式,分别叫做 Locality Preserving Projection 和 Neighborhood Preserving Embedding 。
在 LPP 中,降维函数跟 PCA 中一样被定为是一个线性变换,用一个降维矩阵?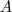 ?来表示,于是 LE 的目标函数变为
?来表示,于是 LE 的目标函数变为
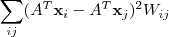
经过类似的推导,最终要求解的特征值问题如下:
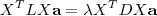
得到的按照特征值从小到大排序的特征向量就组成映射矩阵?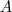 ?,和 LE 不同的是这里不需要去掉第一个特征向量。另一点是在 LE 中的特征值是一个稀疏的特征值问题,在只需要求解最小的几个特征值的时候可以比较高效地求解,而这里的矩阵在乘以?
?,和 LE 不同的是这里不需要去掉第一个特征向量。另一点是在 LE 中的特征值是一个稀疏的特征值问题,在只需要求解最小的几个特征值的时候可以比较高效地求解,而这里的矩阵在乘以?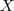 ?之后通常就不再稀疏了,计算量会变得比较大,这个问题可以使用 Spectral Regression 的方法来解决,参见?Spectral Regression: A Unified Approach for Sparse Subspace Learning?这篇 paper 。如果采用 Kernel Trick 再把 LPP 非线性化的话,又会回到 LE 。而 LLE 的线性版本 NPE 也是采用了类似的办法来得到的,就不在这里多讲了。
?之后通常就不再稀疏了,计算量会变得比较大,这个问题可以使用 Spectral Regression 的方法来解决,参见?Spectral Regression: A Unified Approach for Sparse Subspace Learning?这篇 paper 。如果采用 Kernel Trick 再把 LPP 非线性化的话,又会回到 LE 。而 LLE 的线性版本 NPE 也是采用了类似的办法来得到的,就不在这里多讲了。
另外,虽然 LE 是 unsupervised 的,但是如果训练数据确实有标签可用,也是可以加以利用的——在构造相似度矩阵的时候,属于同一类别的相似度要大一些,而不同类别的相似度则会小一些。
当然,除去聚类或分类之外,降维本身也是一种比较通用的数据分析的方法,不过有许多人批评降维,说得到的结果没有意义,不用说非线性,就是最简单的线性降维,除去一些非藏极端的特殊情况的话,通常将原来的分量线性组合一下都不会得到什么有现成的物理意义的量了。然而话也说回来,现在的机器学习几乎都是更 prefer "黑盒子"式的方法吧,比如决策树,各个分支对应与变量的话,它的决策过程其实人是可以"看到"或者说"理解"的,但是 SVM 就不那么"直观"了,如果再加上降维处理,就更加"不透明"了。不过我觉得这没什么不好的,如果只是靠可以清晰描诉出来的 rule 的话,似乎感觉神秘感不够,没法发展出"智能"来啊 ^_^bb 最后,所谓有没有物理意义,其实物理量不过也都是人为了描述问题方便而定义出来的吧。
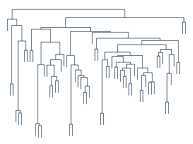 本系列不小心又拖了好久,其实正儿八经的 blog 也好久没有写了,因为比较忙嘛,不过觉得 Hierarchical Clustering 这个话题我能说的东西应该不多,所以还是先写了吧(我准备这次一个公式都不贴?)。Hierarchical Clustering 正如它字面上的意思那样,是层次化的聚类,得出来的结构是一棵树,如右图所示。在前面我们介绍过不少聚类方法,但是都是"平坦"型的聚类,然而他们还有一个更大的共同点,或者说是弱点,就是难以确定类别数。实际上,(在某次不太正式的电话面试里)我曾被问及过这个问题,就是聚类的时候如何确定类别数。
本系列不小心又拖了好久,其实正儿八经的 blog 也好久没有写了,因为比较忙嘛,不过觉得 Hierarchical Clustering 这个话题我能说的东西应该不多,所以还是先写了吧(我准备这次一个公式都不贴?)。Hierarchical Clustering 正如它字面上的意思那样,是层次化的聚类,得出来的结构是一棵树,如右图所示。在前面我们介绍过不少聚类方法,但是都是"平坦"型的聚类,然而他们还有一个更大的共同点,或者说是弱点,就是难以确定类别数。实际上,(在某次不太正式的电话面试里)我曾被问及过这个问题,就是聚类的时候如何确定类别数。
我能想到的方法都是比较 naive 或者比较不靠谱的,比如:
当时对方问"你还有没有什么问题"的时候我竟然忘记了问他这个问题到底有没有什么更好的解决办法,事后真是相当后悔啊。不过后来在实验室里询问了一下,得到一些线索,总的来说复杂度是比较高的,待我下次有机会再细说(先自己研究研究)。
不过言归正传,这里要说的 Hierarchical Clustering 从某种意义上来说也算是解决了这个问题,因为在做 Clustering 的时候并不需要知道类别数,而得到的结果是一棵树,事后可以在任意的地方横切一刀,得到指定数目的 cluster ,按需取即可。
听上去很诱人,不过其实 Hierarchical Clustering 的想法很简单,主要分为两大类:agglomerative(自底向上)和 divisive(自顶向下)。首先说前者,自底向上,一开始,每个数据点各自为一个类别,然后每一次迭代选取距离最近的两个类别,把他们合并,直到最后只剩下一个类别为止,至此一棵树构造完成。
看起来很简单吧???其实确实也是比较简单的,不过还是有两个问题需要先说清除才行:
总的来说,一般都不太用 Single Linkage 或者 Complete Linkage 这两种过于极端的方法。整个 agglomerative hierarchical clustering 的算法就是这个样子,描述起来还是相当简单的,不过计算起来复杂度还是比较高的,要找出距离最近的两个点,需要一个双重循环,而且 Group Average 计算距离的时候也是一个双重循环。
另外,需要提一下的是本文一开始的那个树状结构图,它有一个专门的称呼,叫做?Dendrogram,其实就是一种二叉树,画的时候让子树的高度和它两个后代合并时相互之间的距离大小成比例,就可以得到一个相对直观的结构概览。不妨再用最开始生成的那个三个 Gaussian Distribution 的数据集来举一个例子,我采用 Group Average 的方式来计算距离,agglomerative clustering 的代码很简单,没有做什么优化,就是直接的双重循环:
| ? ? |
数据点又一千多个,画出来的 dendrogram 非常大,为了让结果看起来更直观一点,我把每个叶节点用它本身的 label 来染色,并且向上合并的时候按照权重混合一下颜色,最后把图缩放一下得到这样的一个结果(点击查看原图):
或者可以把所有叶子节点全部拉伸一下看,在右边对齐,似乎起来更加直观一点:

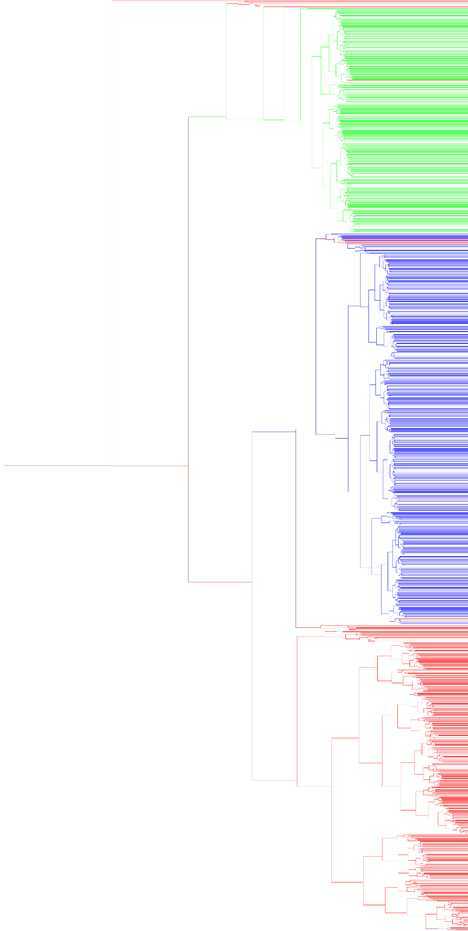
从这个图上可以很直观地看出来聚类的结果,形成一个层次,而且也在总体上把上个大类分开来了。由于这里我把图横过来画了,所以在需要具体的 flat cluster 划分的时候,直观地从图上可以看成竖着划一条线,打断之后得到一片"森林",再把每个子树里的所有元素变成一个"扁平"的集合即可。完整的 Python 代码如下:
| ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? ? |
agglomerative clustering 差不多就这样了,再来看 divisive clustering ,也就是自顶向下的层次聚类,这种方法并没有 agglomerative clustering 这样受关注,大概因为把一个节点分割为两个并不如把两个节点结合为一个那么简单吧,通常在需要做 hierarchical clustering 但总体的 cluster 数目又不太多的时候可以考虑这种方法,这时可以分割到符合条件为止,而不必一直分割到每个数据点一个 cluster 。
总的来说,divisive clustering 的每一次分割需要关注两个方面:一是选哪一个 cluster 来分割;二是如何分割。关于 cluster 的选取,通常采用一些衡量松散程度的度量值来比较,例如 cluster 中距离最远的两个数据点之间的距离,或者 cluster 中所有节点相互距离的平均值等,直接选取最"松散"的一个 cluster 来进行分割。而分割的方法也有多种,比如,直接采用普通的 flat clustering 算法(例如 k-means)来进行二类聚类,不过这样的方法计算量变得很大,而且像 k-means 这样的和初值选取关系很大的算法,会导致结果不稳定。另一种比较常用的分割方法如下:
到此为止,我的 hierarchical clustering 介绍就结束了。总的来说,在我个人看来,hierarchical clustering 算法似乎都是描述起来很简单,计算起来很困难(计算量很大)。并且,不管是 agglomerative 还是 divisive 实际上都是贪心算法了,也并不能保证能得到全局最优的。而得到的结果,虽然说可以从直观上来得到一个比较形象的大局观,但是似乎实际用处并不如众多 flat clustering 算法那么广泛。
如何确定聚类的类别个数这个问题经常有人问我,也是一直以来让我自己也比较困惑的问题。不过说到底其实也没什么困惑的,因为这个问题本身就是一个比较 ill posed 的问题呀:给一堆离散的点,要你给出它们属于几个 cluster,这个基本上是没有唯一解或者说是没有合适的标准来衡量的。比如如果简单地用每个类别里的点到类中心的距离之和来衡量的话,一下子就会进入到"所有的点都独立成一类"这样的尴尬境界中。
但是 ill posed 也并不是一个很好的理由,因为我们其实大部分时候都是在处理 ill posed 的问题嘛,比如 Computer Vision 整个一个领域基本上就没有啥问题是 well posed 的……?=.=bb,比如下面盗用一下?Bill Freeman?的?Slides?中的一张讲 deblur 的问题的图:
Blurry image Sharp image Blur kernel
| ? | = |
|
| |
|
|
| |||
= |
|
|
从 Stata Center 的模糊照片找出清晰照片和模糊核的这个过程(特别对于计算机来说)就是非常 ambigous 的。(顺便这个?Slides?本身也是很有意思的,推荐看一下。)
所以么,还是让我们先抛开各种借口,回到问题本身。当然这次的标题取得有点宏大,其实写这篇日志的主要目的不过是想吐槽一下上一次 6.438 课的一道作业题而已……=.=bb
总而言之呢,像?Kmeans?之类的大部分经典的聚类算法,都是需要事先指定一个参数K作为类别数目的。但是很多时候这个K值并不是那么好确定的,更麻烦的是,即使你使用很暴力的方法把某个范围内的整数K全部都试一遍,通常也没法知道哪个K才是最好的。
所以呢,我们自然会问:那有没有算法不需要指定类别数呢?当然有!最简单的就是层次聚类,它会将你的数据点用一个树形结构给连起来,可是我想要的是聚类结果啊!这也好办,只要在合适的树深度的地方把树切开,变成一个一个的子树,每个子树就是一个cluster。不过问题就来了,究竟什么深度才是合适的深度呢?事实上不同的深度会产生不同的类别数目,这基本上把原来的选择K的问题转化成了选择树深度的问题。
没关系,我们还有其他货,比如著名的?Mean Shift?算法,也是不需要指定类别数目就可以聚类的,而且聚类的结果也不是一棵树那种奇奇怪怪的东西。具体 Mean Shift 算法是什么样的今天就不在这里讲了,不过它其实也有一个参数 Window Size 需要选择——对,正如大家所料,这个 Window Size 其实也是会影响最终聚类的类别数目。换句话说,原来选择K的问题现在被转化为了选择 Window Size 的问题。
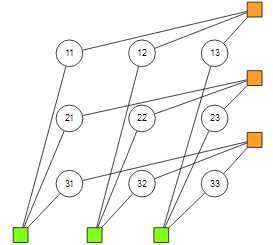
图 2????Factor graph demonstration for Affinity Propagation of 3 points.
然后我们进入贝叶斯推断的领域,之前跟别人聊天的时候就听说过有个叫做 Dirichlet Process 的东西,特别神奇,能够自动地根据数据 adaptive 地确定合适的类别数目。我感觉这背后可能像其他算法一样会隐藏着其他的参数需要调节,不过鉴于我对 DP 完全不懂,就不在这里说胡话了,感兴趣的同学可以去研究一下。
然后贝叶斯推断聚类里的另一个成员就是 Affinity Propagation,其实是在讲了 Loopy Belief Propagation 算法之后的一道作业题,给了一个 factor graph,让把消息传递的公式推出来然后实现出来。看起来好像蛮容易的样子,实际上折腾了好几天,因为实现的时候碰到各种 tricky 的问题。
AP 算法把聚类问题看成一个 MAP 推断问题,假设我们有n个数据点,这里我们要求类别中心实际上是这些数据点中的一个子集,聚类的结果可以用这个binary variable 来表示,其中表示点被归到点所代表的那个类别中。
给定个随机变量之后,接下来是要通过 local factor 来定义他们的 (unnormalized) joint distribution。首先我们要求已知一个 Affinity Matrix?S,其中表示点和点之间的相似程度(这里??并不一定要求是对称的),一个简单的做法就是令
然后整个 joint distribution 主要由两类 factor 构成,在图?(fig: 2)?中给出了 3 个数据点时候的 factor graph 的例子。其中橙色的 factor 定义为
直观地来讲,该 factor 的第一部分是说每个点只能且必须被 assign 到一个 cluster;第二部分是讲被assign 到的时候 factor 的值为?,也就是说会偏向于 assign 到相似度较大的那个 cluster 去。
而绿色的那些 factor 则定义为:
这样的 indicator factor 要求如果有任何点被 assign 到的话,那么必须要被 assign 到它自己。换句话说,每个 cluster 的中心必须是属于它自己那个类的,这也是非常合理的要求。
好了,模型的定义就是这么简单,这样我们会得到一个 loopy factor graph,接下来只要在这个 graph 上跑一下 Max-Product Algorithm 就可以得到最优的?,从而得到聚类结果了,并且都不用指定类别数目什么的,因为合适的类别数目可以从数据中自动地推断出来!
简直太神奇了,我都不敢相信这是真的!结果,果然童话里都是骗人的……因为隐藏在背后的还有许多细节需要处理。首先有一些 trick 来降低算法的复杂程度,比如说,由于所有的变量都是 binary 的,因此在消息传递的过程中我们可以只传递等于 1 和等于 0 两种状态时的消息差;然后由于数值下溢的原因,一般都会对所有的消息取log?,这样 Max-Product 算法会变成 Max-Sum(或者 Min-Sum);然后有些消息是不做任何操作就直接 literally 传到接下来的节点的,这样的消息可以从中间步骤中去掉,最后化简之后的结果会只留下两种消息,在经典的 Affinity Propagation 算法中分别被成为 responsibility 消息和 availability 消息,可以有另外的 intuitive 的解释,具体可以参见?(Frey & Dueck, 2007)。

 图 3????Clustering result of Affinity Propagation.
图 3????Clustering result of Affinity Propagation.
不过这些还是不太够,因为 Loopy BP 的收敛性其实是没有什么保证的,随便实现出来很可能就不收敛。比如说,如果直接进行 parallel 地 message updating 的话,很可能就由于 update 的幅度过大导致收敛困难,因此需要用 dampen 的方法将 fixed-point 的式子
改为
然后用这个来做 updating。当然也可以用 0.5 以外的其他 dampen factor。不过这样还是不够的,反正我折腾了好久最后发现必须把 parallel updating 改成 in-place updating 才能收敛,这样一来更新的幅度就更小了一些,不过 dampen 仍然是需要的。另外就是一同讨论的另外的同学还发现实现中消息更新的顺序也是极端重要……把顺序变一下就立马从完全不 work 变成结果超好了。-_-!!于是突然想起来 CV 课上老师拿 Convolutional Network 开玩笑的时候说,这个算法效果超级好,但是就是太复杂了,在发明这个算法的那个 lab 所在地的方圆 50 米之外完全没人能成功训练 Convolutional Network……

 图 4????Clustering result of Affinity Propagation, with large diagonal values for the affinity matrix.
图 4????Clustering result of Affinity Propagation, with large diagonal values for the affinity matrix.
此外收敛性的判断也是有各种方法,比如消息之差的绝对值、或者是连续多少轮迭代产生的聚类中心都没有发生变化等等。即使在收敛之后,如何确定 assignment 也并不是显而易见的。因为 Max-Product 算法得到的是一些 Max-Marginal,如果全局最优的 configuration 不是 unique 的话,general 地是不能直接局部地通过各自的 Max-Marginal 来确定全局 configuration 的,于是可能需要实现 back-tracking,总之就是动态规划的东西。不过也有简单的 heuristic 方法可以用,比如直接通过的Max-Marginal 来确定点到底是不是一个 cluster center,确定了 center 之后则再可以通过 message 或者直接根据 Affinity 矩阵来确定最终的类别 Assignment。图?(fig: 3)?展示了一个二维情况下 100 个点通过 AP 聚类的结果。
不过,你有没有发现一点不对劲的地方?一切似乎太完美了:什么参数都不用给,就能自动确定类别数?果然还是有问题的。事实上,如果你直接就去跑这个算法,肯定得不到图上的结果,而是会得到一个n个类别的聚类结果:所有的点都成为了中心并且被归类到了自己。这样的结果也是可以理解的,因为我们计算 Affinity 的方式(距离的相反数)导致每个点和自己的 Affinity 是最大的……所以呢,为了解决这个问题,我们需要调整一个参数,就是S矩阵的对角线上的值,这个值反应了每个点自己成为类别中心的偏好程度,对的,如大家所料,这个就是背后的隐藏参数,直接影响类别数目。

 图 5????Clustering result of Affinity Propagation, with small diagonal values for the affinity matrix.
图 5????Clustering result of Affinity Propagation, with small diagonal values for the affinity matrix.
如图?(fig: 4)?和?(fig: 5)?分别是把对角线上的元素值设置得比较大和比较小的情况,可以看到前者产生了更多的类别,因为大家都比较倾向于让自己成为类别中心;而后者则只产生了两个类,因为大家都很不情愿……(我发现这种画一些线连到类别中心的 visualization 方法似乎会给人产生很强的暗示感觉,于是各种聚类结果都看起来好像很"对"的样子)
所以呀,到最后好像还是竹篮打水一场空的样子,怎么绕怎么绕好像也绕不开类别数目的这个参数。不过好像也并不是完全没有任何进展的,因为虽然各种算法都或多或少地需要指定一些参数,但是这些参数从不同的角度来阐释对应的问题,比如说在 AP 中,一般可以直接将??的对角线元素设置为其他所以元素的中位数,通常就能得到比较合理的结果;有时候对于特定的实际问题来说,从某一个角度来进行参数选择可能会有比较直观的 heuristic 可以用呢。所以说在是否需要指定参数这个问题上,还是不用太钻牛角尖了啊。
最后我还想提一下,最近看到的 lab 的两篇 NIPS 文章?(Canas, Poggio & Rosasco, 2012),(Canas & Rosasco, 2012),里面的视角也是很有意思的。
我们知道,比如说,KMeans 算法的目标函数是这样定义的:
其中是一个由K个点组成的集合,而一个点到的距离被定义为到S中所有点的距离中最小的那一个值。这样可以看成是用的这K个点去近似原本的n个点所带来的误差。然而这样的目标函数对于选择合适的K并没有什么指导意义,因为随着K的增大我们得到的最优的会使得目标函数越来越小。
但是我们可以将这个目标函数推广一下,类似于 Supervised Learning Theory 中那样引入概率空间。具体地来说,我们假设数据点是从一个未知的概率分布中采样而得到的,并且目标函数也从对n个数据点的近似推广到整个生成流形的近似:
当然由于是未知的,所以是没法计算的,但是我们可以从 data sample 得到的来对它进行近似,并得到 bound 之类的。直观地来讲,此时对数值K的选择将会影响到模型空间的复杂度,于是会出现一个trade-off,于是从这个角度下去探讨"最优的K值"就变成一个很合理的问题了。感兴趣的同学可以详细参考 paper 以及里面的参考文献,都在 arXiv 上可以下载到的。
References
标签:des style blog http io color ar os 使用
原文地址:http://www.cnblogs.com/CVFans/p/4103430.html