最近在做组装稻瘟病的基因组。 估计的基因组大小为40M。 由于没有参考基因组,进行de novo assembly。 用HGAP策略。需要的有用的pacbio数据量应为400M左右, 选用的seed read 最小长度为6K, seed的覆盖度应在20倍左右。
拼接流程为:1,filtering. 2, assembly. 3, mapping. 4, consensus.选用的cutoff如下图:
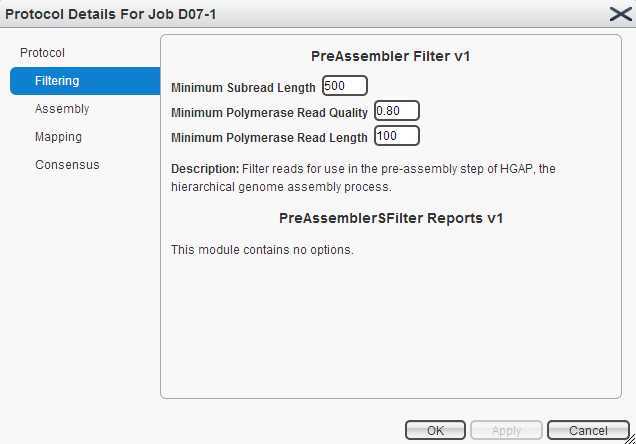
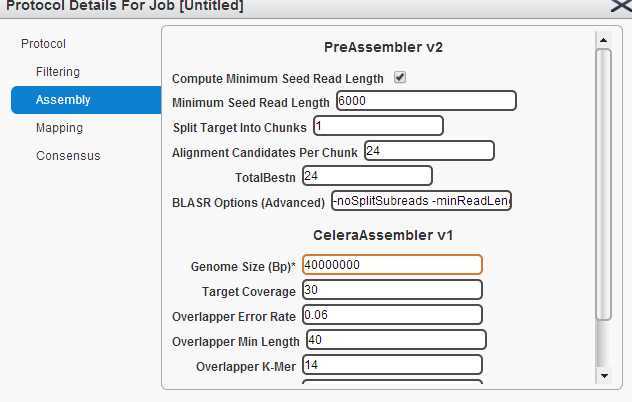
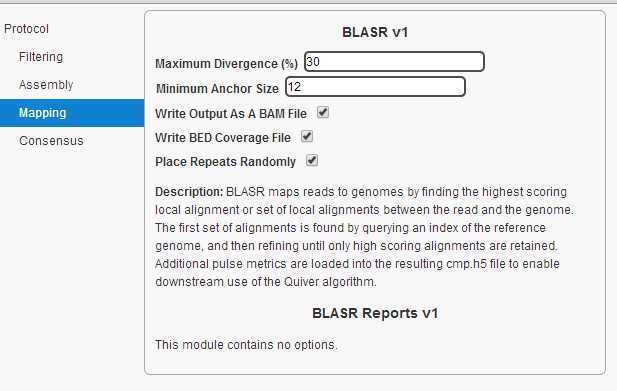
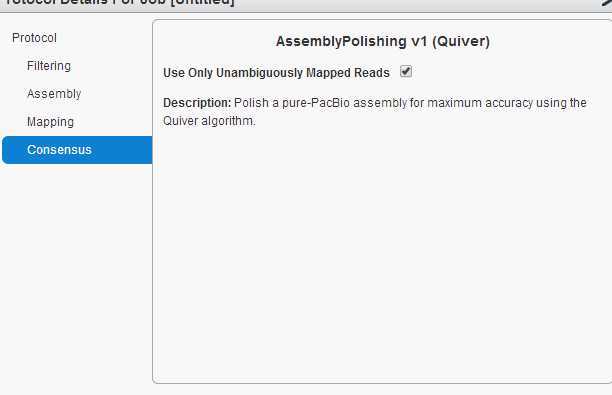
所有的操作都在网页上进行。 piobio 的数据以cell 为单位。每个cell中有很多ZMWS, ZMWS可以产生三种产物。
productivity0: ZMWS中没有聚合酶,相当于是空的
productivity1:有聚合酶存在,产生了有效的数据。
productivity2:虽然不是空的,但是产生的数据是不能用的。
所以,对于每个cell来说,只有productivity1 是有用的。
首先导入你的数据,1,DESIGN JOB 2, Import and Manage 3, from smrt cell 4, 添加你的cell 数据在服务器中的位置,添加好后,scan, 如果数据正确,就会被导入。
注意: 对于每一个cell来说,必须有Analysis_Results文件夹, 其中metadata.xml和Analysis_Results在同一个目录下。bas.h5 和bax.h5在Analysis_Results目录下。 如果目录结构不正确是不能导入该cell的。bax.h5文件必须有,另外的没有测试过。
将所有的cell导入后,就可以建立一个job了。1, DESIGN JOB 2,Creat New 3, 填写job name ,comments 是关于这个job的说明,可写可不写。选择protocol, 由于是de novo assembly,选择RS_HGAP_Assembly.2。这个protocol的设置如上面的图所示。将属于该job的cell数据导入, 所以,你必须知道每个cell的ID, ID如何查看直接看原始数据就知道了。这些做完以后,点击save, start.job就开始跑了。
跑完以后会生成此次job的报告, 你可以根据报告查看这次job的情况。最终的结果位置可以在log文件中找到(eg:/opt/smrtanalysis/install/smrtanalysis-2.1.1.128549/common/jobs/016/016451)。出现错误的话log文件也会记录出错信息。
关于HGAP 组装策略 请自己下载参考文献阅读, 这里不多说明。
by freemao
FAFU.
free_mao@qq.com
利用 pacbio 数据组装真菌基因组,布布扣,bubuko.com
原文地址:http://www.cnblogs.com/freemao/p/3783475.html